Concurrently mapping quantitative trait loci associations from multiple subspecies within hybrid populations
IF 3.1
2区 生物学
Q2 ECOLOGY
引用次数: 0
Abstract
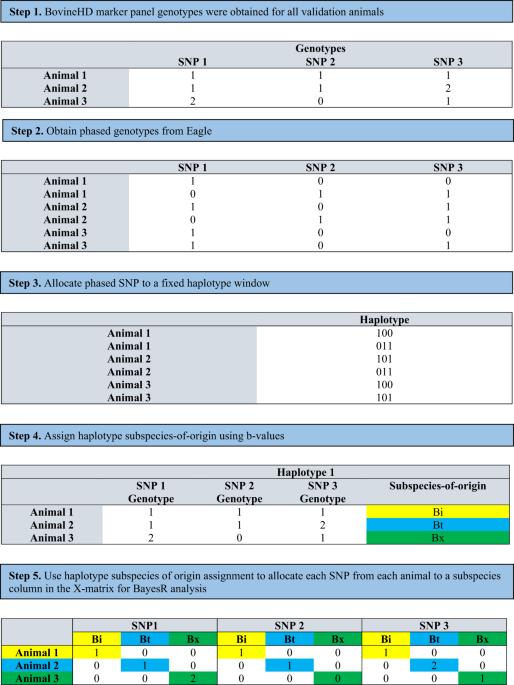
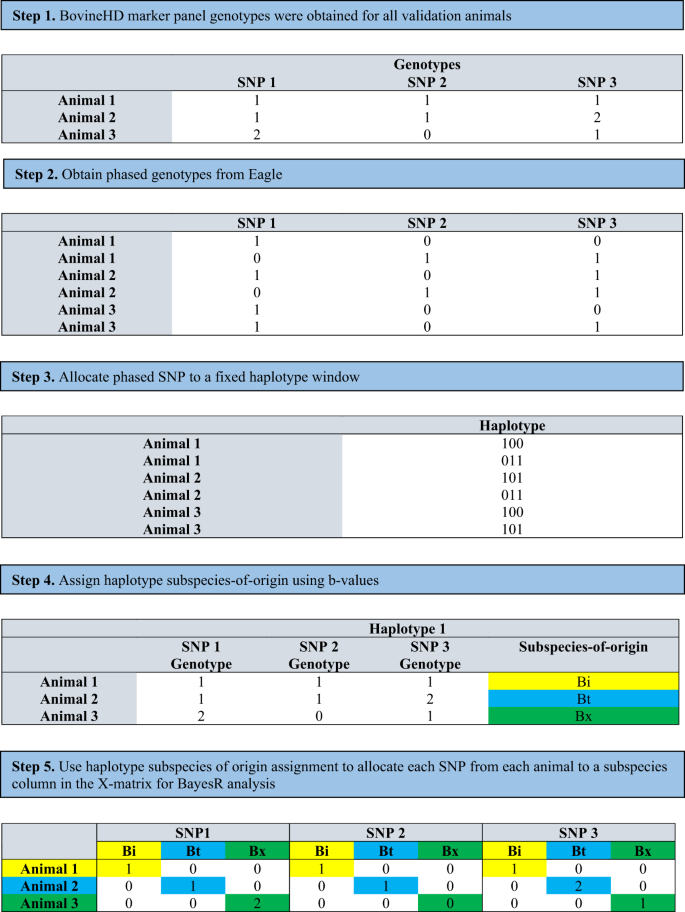
Many of the world’s agriculturally important plant and animal populations consist of hybrids of subspecies. Cattle in tropical and sub-tropical regions for example, originate from two subspecies, Bos taurus indicus (Bos indicus) and Bos taurus taurus (Bos taurus). Methods to derive the underlying genetic architecture for these two subspecies are essential to develop accurate genomic predictions in these hybrid populations. We propose a novel method to achieve this. First, we use haplotypes to assign SNP alleles to ancestral subspecies of origin in a multi-breed and multi-subspecies population. Then we use a BayesR framework to allow SNP alleles originating from the different subspecies differing effects. Applying this method in a composite population of B. indicus and B. taurus hybrids, our results show that there are underlying genomic differences between the two subspecies, and these effects are not identified in multi-breed genomic evaluations that do not account for subspecies of origin effects. The method slightly improved the accuracy of genomic prediction. More significantly, by allocating SNP alleles to ancestral subspecies of origin, we were able to identify four SNP with high posterior probabilities of inclusion that have not been previously associated with cattle fertility and were close to genes associated with fertility in other species. These results show that haplotypes can be used to trace subspecies of origin through the genome of this hybrid population and, in conjunction with our novel Bayesian analysis, subspecies SNP allele allocation can be used to increase the accuracy of QTL association mapping in genetically diverse populations.
同时绘制杂交种群中多个亚种的数量性状基因座关联图。
世界上许多在农业上重要的动植物种群都由亚种的杂交种组成。例如,热带和亚热带地区的牛起源于两个亚种,即印度野牛(Bos indicus)和美洲野牛。推导这两个亚种潜在遗传结构的方法对于在这些杂交种群中进行准确的基因组预测至关重要。我们提出了一种新的方法来实现这一点。首先,我们使用单倍型将SNP等位基因分配给多品种和多亚种群体中的祖先亚种。然后,我们使用贝叶斯R框架来允许来自不同亚种的SNP等位基因产生不同的影响。将该方法应用于印度牛和牛头牛杂交种的复合种群,我们的结果表明,这两个亚种之间存在潜在的基因组差异,并且在不考虑起源亚种影响的多品种基因组评估中没有发现这些影响。该方法略微提高了基因组预测的准确性。更重要的是,通过将SNP等位基因分配给祖先的起源亚种,我们能够识别出四个具有高后验概率的SNP,这些SNP以前与牛的生育能力无关,并且与其他物种的生育能力相关的基因接近。这些结果表明,单倍型可用于通过该杂交群体的基因组追踪起源亚种,结合我们新的贝叶斯分析,亚种SNP等位基因分配可用于提高遗传多样性群体中QTL关联图谱的准确性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Heredity
生物-进化生物学
CiteScore
7.50
自引率
2.60%
发文量
84
审稿时长
4-8 weeks
期刊介绍:
Heredity is the official journal of the Genetics Society. It covers a broad range of topics within the field of genetics and therefore papers must address conceptual or applied issues of interest to the journal''s wide readership
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


