{"title":"Changes in uridine 5′-diphospho-glucuronosyltransferase 1A6 expression by histone deacetylase inhibitor valproic acid","authors":"Yukiko Sakakibara, Ayaka Kojima, Yuki Asai, Masayuki Nadai, Miki Katoh","doi":"10.1002/bdd.2328","DOIUrl":null,"url":null,"abstract":"<p>Valproic acid (VPA) is well-known as a histone deacetylase (HDAC) inhibitor. It has been reported that HDAC inhibitors enhance basal and aryl hydrocarbon receptor (AhR) ligand-induced aryl hydrocarbon receptor-responsive gene expression. Other studies suggested that HDAC inhibition might significantly activate the NF-E2-related factor-2 (Nrf2). Moreover, VPA activates mitogen-activated protein kinases (MAPKs). MAPK pathways regulate Nrf2 transactivation domain activity. Uridine 5′-diphospho-glucuronosyltransferase (UGT) 1A6 is one of the important isoforms to affect drug pharmacokinetics. UGT1A6 gene is regulated transcriptionally by AhR and Nrf2. The present study aimed to investigate whether UGT1A6 expression was changed by VPA and to elucidate the mechanism of the alteration. Following VPA treatment for 72 h in Caco-2 cells, UGT1A6 mRNA was increased by 7.9-fold. Moreover, UGT1A6 mRNA was increased by other HDAC inhibitors, suggesting that HDAC inhibition caused the UGT1A6 mRNA induction. AhR and Nrf2 proteins in the nucleus of Caco-2 cells were increased by 1.5- and 1.7-fold, respectively, following the VPA treatment. However, VPA treatment did not activate the extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways in Caco-2 cells. In conclusion, we observed that VPA induced UGT1A6 mRNA expression via AhR and Nrf2 pathways, but not via the ERK or JNK pathways.</p>","PeriodicalId":8865,"journal":{"name":"Biopharmaceutics & Drug Disposition","volume":"43 5","pages":"175-182"},"PeriodicalIF":2.0000,"publicationDate":"2022-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biopharmaceutics & Drug Disposition","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bdd.2328","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 1
Abstract
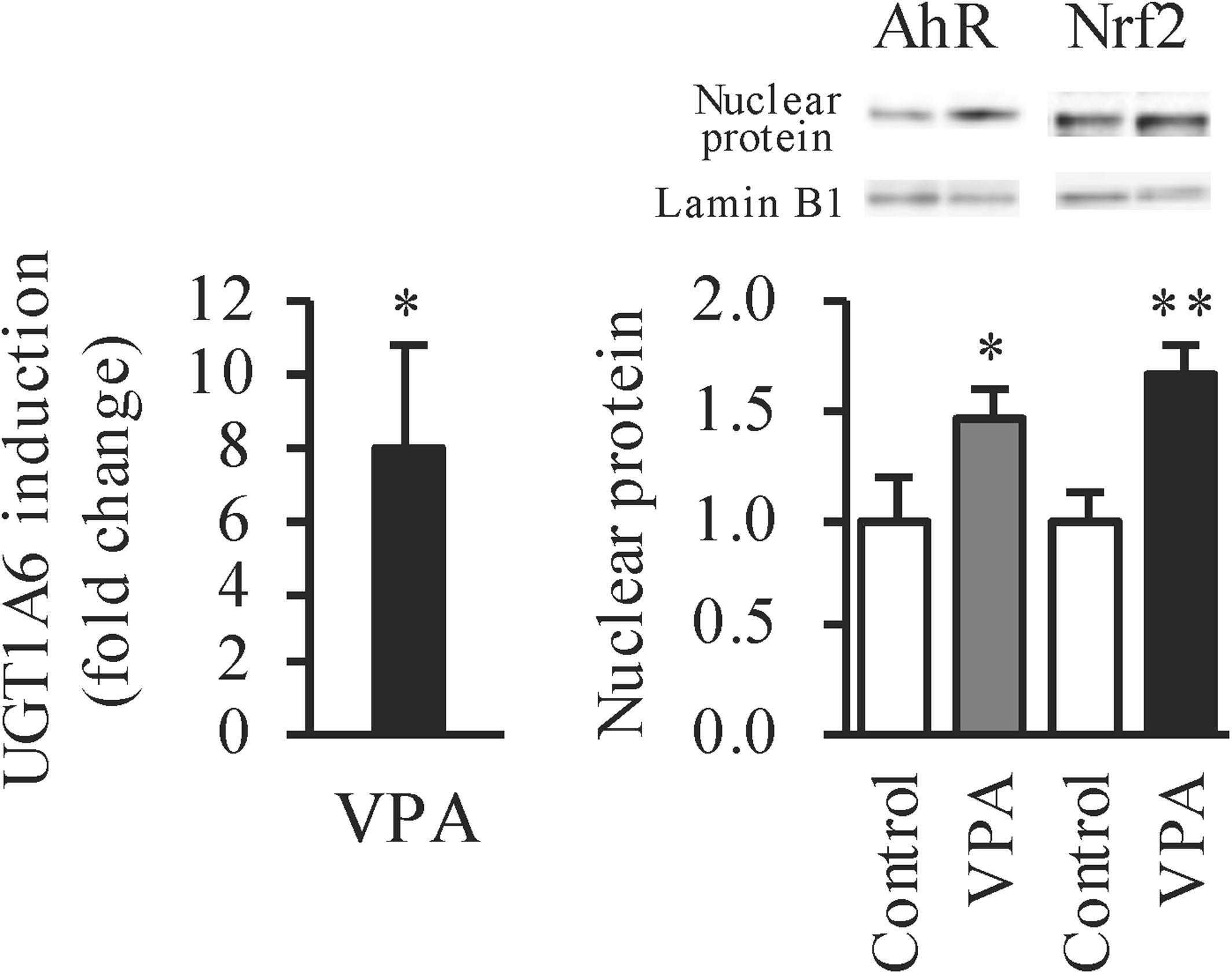
Valproic acid (VPA) is well-known as a histone deacetylase (HDAC) inhibitor. It has been reported that HDAC inhibitors enhance basal and aryl hydrocarbon receptor (AhR) ligand-induced aryl hydrocarbon receptor-responsive gene expression. Other studies suggested that HDAC inhibition might significantly activate the NF-E2-related factor-2 (Nrf2). Moreover, VPA activates mitogen-activated protein kinases (MAPKs). MAPK pathways regulate Nrf2 transactivation domain activity. Uridine 5′-diphospho-glucuronosyltransferase (UGT) 1A6 is one of the important isoforms to affect drug pharmacokinetics. UGT1A6 gene is regulated transcriptionally by AhR and Nrf2. The present study aimed to investigate whether UGT1A6 expression was changed by VPA and to elucidate the mechanism of the alteration. Following VPA treatment for 72 h in Caco-2 cells, UGT1A6 mRNA was increased by 7.9-fold. Moreover, UGT1A6 mRNA was increased by other HDAC inhibitors, suggesting that HDAC inhibition caused the UGT1A6 mRNA induction. AhR and Nrf2 proteins in the nucleus of Caco-2 cells were increased by 1.5- and 1.7-fold, respectively, following the VPA treatment. However, VPA treatment did not activate the extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways in Caco-2 cells. In conclusion, we observed that VPA induced UGT1A6 mRNA expression via AhR and Nrf2 pathways, but not via the ERK or JNK pathways.
期刊介绍:
Biopharmaceutics & Drug Dispositionpublishes original review articles, short communications, and reports in biopharmaceutics, drug disposition, pharmacokinetics and pharmacodynamics, especially those that have a direct relation to the drug discovery/development and the therapeutic use of drugs. These includes:
- animal and human pharmacological studies that focus on therapeutic response. pharmacodynamics, and toxicity related to plasma and tissue concentrations of drugs and their metabolites,
- in vitro and in vivo drug absorption, distribution, metabolism, transport, and excretion studies that facilitate investigations related to the use of drugs in man
- studies on membrane transport and enzymes, including their regulation and the impact of pharmacogenomics on drug absorption and disposition,
- simulation and modeling in drug discovery and development
- theoretical treatises
- includes themed issues and reviews
and exclude manuscripts on
- bioavailability studies reporting only on simple PK parameters such as Cmax, tmax and t1/2 without mechanistic interpretation
- analytical methods

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


