Zachariah Chowdhury, Yookarin Khonglah, Vandana Raphael, Pranjal Kalita, Umesh Das
{"title":"Hepatosplenic T Cell Lymphoma: Diagnostic Conundrum.","authors":"Zachariah Chowdhury, Yookarin Khonglah, Vandana Raphael, Pranjal Kalita, Umesh Das","doi":"10.18502/ijhoscr.v16i1.8444","DOIUrl":null,"url":null,"abstract":"<p><p>Hepatosplenic T cell lymphoma (HSTCL) is a very rare and aggressive peripheral T cell lymphoma that comprises less than 1% of Non-Hodgkin lymphomas (NHL). It is derived from cytotoxic T-cells, usually of γδ T cell receptor type, and is characterized by primary extranodal disease with typical sinusoidal infiltration of the liver, spleen and bone marrow by medium-sized lymphoid cells. HSTCL occurs more frequently in immunocompromised patients, especially in those receiving long-term immunosuppressive therapy. The differential diagnosis is varied, and the clinical course is dismal with a poor response to currently available therapies. Herein we report a case of HSTCL in a 20-year-old immunocompetent male who presented with fever, pallor, weight loss, bicytopenia, hepatomegaly, and massive splenomegaly, highlighting the diagnostic conundrum and pointers towards an accurate diagnosis. The key role for diagnosis was the combination of morphologic finding of atypical lymphoid cells in the bone marrow, typical immunophenotypic profile on flow cytometry and the pattern of involvement of the liver and the spleen, even in the absence of full-fledged diagnostic panels and tools. The report of this case is an endeavor to emphasize the high index of suspicion for timely detection of such a rare entity.</p>","PeriodicalId":38991,"journal":{"name":"International Journal of Hematology-Oncology and Stem Cell Research","volume":"16 1","pages":"66-73"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ef/4a/IJHOSCR-16-66.PMC9339126.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Hematology-Oncology and Stem Cell Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.18502/ijhoscr.v16i1.8444","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 2
Abstract
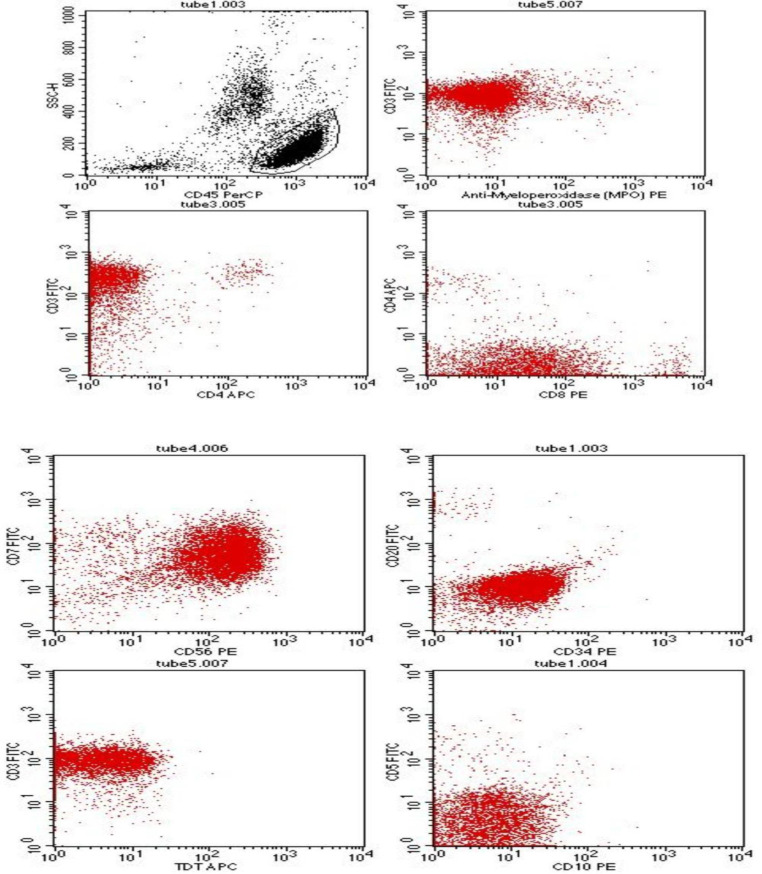
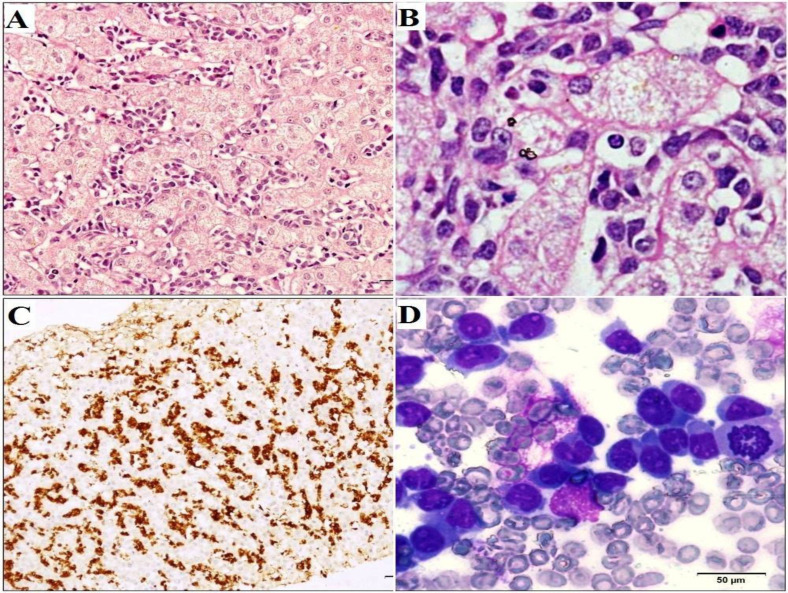
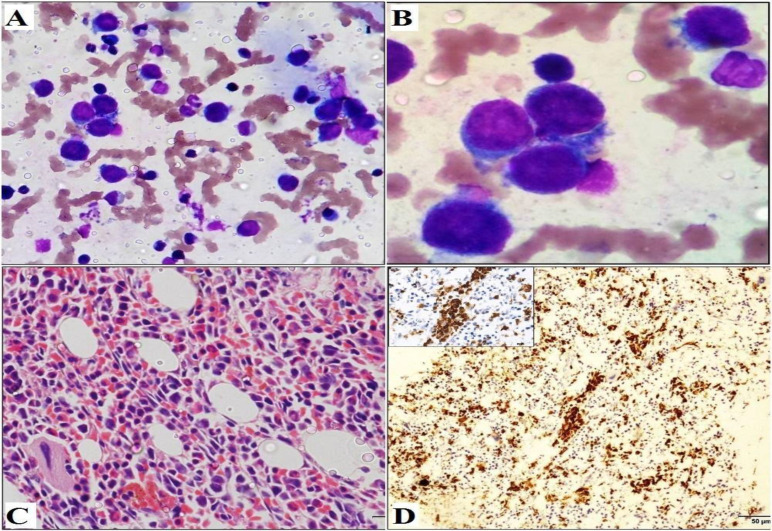
Hepatosplenic T cell lymphoma (HSTCL) is a very rare and aggressive peripheral T cell lymphoma that comprises less than 1% of Non-Hodgkin lymphomas (NHL). It is derived from cytotoxic T-cells, usually of γδ T cell receptor type, and is characterized by primary extranodal disease with typical sinusoidal infiltration of the liver, spleen and bone marrow by medium-sized lymphoid cells. HSTCL occurs more frequently in immunocompromised patients, especially in those receiving long-term immunosuppressive therapy. The differential diagnosis is varied, and the clinical course is dismal with a poor response to currently available therapies. Herein we report a case of HSTCL in a 20-year-old immunocompetent male who presented with fever, pallor, weight loss, bicytopenia, hepatomegaly, and massive splenomegaly, highlighting the diagnostic conundrum and pointers towards an accurate diagnosis. The key role for diagnosis was the combination of morphologic finding of atypical lymphoid cells in the bone marrow, typical immunophenotypic profile on flow cytometry and the pattern of involvement of the liver and the spleen, even in the absence of full-fledged diagnostic panels and tools. The report of this case is an endeavor to emphasize the high index of suspicion for timely detection of such a rare entity.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


