{"title":"Acute Intermittent Porphyria: Complete Phenotype in a Patient with p.Arg173Trp Variant in Thailand.","authors":"Vachiravit Sriprakoon, Chalisa Ittagornpunth, Nakorn Puapaiboon, Aekasit Bunyahathaipat, Punnapat Piriyanon, Sookkasem Khositseth, Kitiwan Rojnueangnit","doi":"10.12659/AJCR.937695","DOIUrl":null,"url":null,"abstract":"<p><p>BACKGROUND Acute intermittent porphyria (AIP) is a rare genetic disease caused by the deficiency of porphobilinogen deaminase enzyme in the heme synthesis pathway. AIP is passed by autosomal dominant inheritance. Heterozygous pathogenic variants in hydroxymethylbilane synthase (HMBS) are associated with AIP. Multisystemic manifestations of acute neurovisceral features exist, which are quite challenging for diagnosis. Currently, few patients worldwide have been reported with AIP. A small number of reports have been published in Thailand, but none have been confirmed by molecular genetics diagnosis. CASE REPORT A 14-year-old female adolescent presented with severe intermittent abdominal pain, vomiting, seizure, posterior reversible encephalopathy syndrome, syndrome of inappropriate antidiuretic hormone, and muscle weakness, which are all classic phenotypes of an acute AIP attack. The patient received several investigations before AIP was suspected. High levels of urine porphobilinogen, high levels of urine aminolevulinic acid, and a heterozygous known pathogenic variant in HMBS: c.517C>T (p.Arg173Trp) were identified. Therefore, AIP was the definitive diagnosis. Then, Sanger sequencing testing was performed for the patient's family; this variant was found in her father, paternal grandmother, and sister, who were all asymptomatic (latent AIP). After the AIP was confirmed, high carbohydrate loading was given as a standard treatment. She had a full recovery; her clinical course of the attack episode lasted for 8 weeks. CONCLUSIONS An early diagnosis of AIP leads to prompt and specific treatment, which can shorten the duration of attacks, prevent complications, reduce the cost of treatment, and reduce the mortality rate.</p>","PeriodicalId":205256,"journal":{"name":"The American Journal of Case Reports","volume":" ","pages":"e937695"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1f/c9/amjcaserep-23-e937695.PMC9641550.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The American Journal of Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12659/AJCR.937695","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
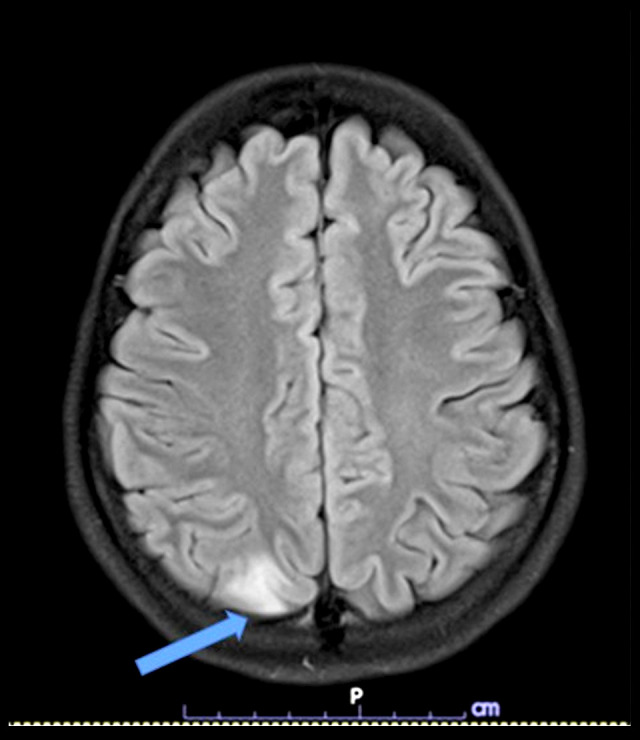
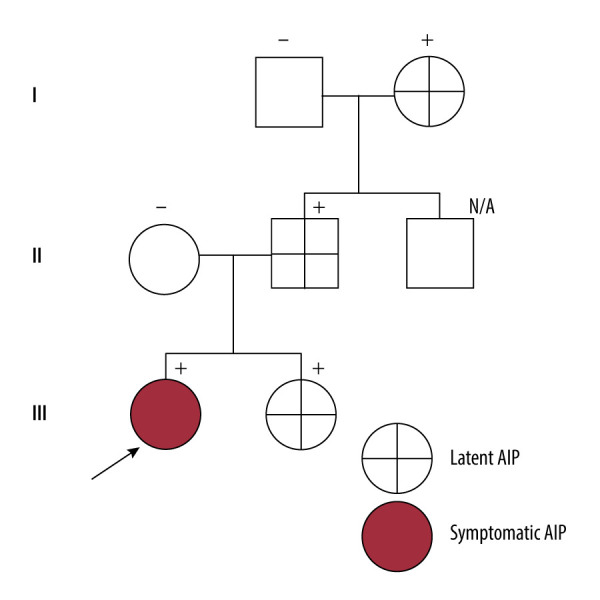
BACKGROUND Acute intermittent porphyria (AIP) is a rare genetic disease caused by the deficiency of porphobilinogen deaminase enzyme in the heme synthesis pathway. AIP is passed by autosomal dominant inheritance. Heterozygous pathogenic variants in hydroxymethylbilane synthase (HMBS) are associated with AIP. Multisystemic manifestations of acute neurovisceral features exist, which are quite challenging for diagnosis. Currently, few patients worldwide have been reported with AIP. A small number of reports have been published in Thailand, but none have been confirmed by molecular genetics diagnosis. CASE REPORT A 14-year-old female adolescent presented with severe intermittent abdominal pain, vomiting, seizure, posterior reversible encephalopathy syndrome, syndrome of inappropriate antidiuretic hormone, and muscle weakness, which are all classic phenotypes of an acute AIP attack. The patient received several investigations before AIP was suspected. High levels of urine porphobilinogen, high levels of urine aminolevulinic acid, and a heterozygous known pathogenic variant in HMBS: c.517C>T (p.Arg173Trp) were identified. Therefore, AIP was the definitive diagnosis. Then, Sanger sequencing testing was performed for the patient's family; this variant was found in her father, paternal grandmother, and sister, who were all asymptomatic (latent AIP). After the AIP was confirmed, high carbohydrate loading was given as a standard treatment. She had a full recovery; her clinical course of the attack episode lasted for 8 weeks. CONCLUSIONS An early diagnosis of AIP leads to prompt and specific treatment, which can shorten the duration of attacks, prevent complications, reduce the cost of treatment, and reduce the mortality rate.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


