In silico Drug Repurposing of Anticancer Drug 5-FU and Analogues Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics Simulation, Pharmacokinetics and Chemical Reactivity Studies.
Aristote Matondo, Washington Dendera, Bienfait Kabuyaya Isamura, Koto-Te-Nyiwa Ngbolua, Hilaire V S Mambo, Mayaliwa Muzomwe, Virima Mudogo
{"title":"In silico Drug Repurposing of Anticancer Drug 5-FU and Analogues Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics Simulation, Pharmacokinetics and Chemical Reactivity Studies.","authors":"Aristote Matondo, Washington Dendera, Bienfait Kabuyaya Isamura, Koto-Te-Nyiwa Ngbolua, Hilaire V S Mambo, Mayaliwa Muzomwe, Virima Mudogo","doi":"10.2147/AABC.S366111","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Since the last COVID-19 outbreak, several approaches have been given a try to quickly tackle this global calamity. One of the well-established strategies is the drug repurposing, which consists in finding new therapeutic uses for approved drugs. Following the same paradigm, we report in the present study, an investigation of the potential inhibitory activity of 5-FU and nineteen of its analogues against the SARS-CoV-2 main protease (3CLpro).</p><p><strong>Material and methods: </strong>Molecular docking calculations were performed to investigate the binding affinity of the ligands within the active site of 3CLpro. The best binding candidates were further considered for molecular dynamics simulations for 100 ns to gain a time-resolved understanding of the behavior of the guest-host complexes. Furthermore, the profile of druggability of the best binding ligands was assessed based on ADMET predictions. Finally, their chemical reactivity was elucidated using different reactivity descriptors, namely the molecular electrostatic potential (MEP), Fukui functions and frontier molecular orbitals.</p><p><strong>Results and discussion: </strong>From the calculations performed, four candidates (compounds 14, 15, 16 and 18) show promising results with respect to the binding affinity to the target protease, 3CLpro, the therapeutic profile of druggability and safety. These compounds are maintained inside the active site of 3CLpro thanks to a variety of noncovalent interactions, especially hydrogen bonds, involving important amino acids such as GLU166, HIS163, GLY143, ASN142, HIS172, CYS145. Molecular dynamics simulations suggest that the four ligands are well trapped within the active site of the protein over a time gap of 100 ns, ligand <b>18</b> being the most retained.</p><p><strong>Conclusion: </strong>In line with the findings reported herein, we recommend that further in-vitro and in-vivo investigations are carried out to shed light on the possible mechanism of pharmacological action of the proposed ligands.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":" ","pages":"59-77"},"PeriodicalIF":0.0000,"publicationDate":"2022-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/da/d5/aabc-15-59.PMC9391940.pdf","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AABC.S366111","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
Abstract
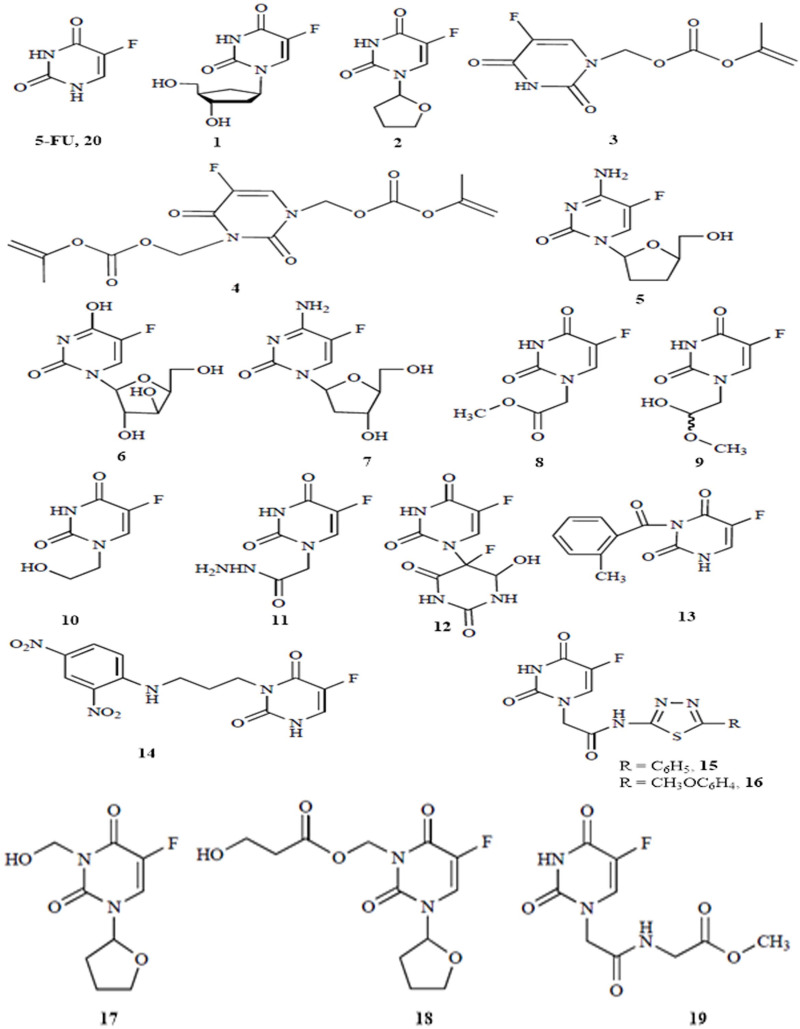
Background: Since the last COVID-19 outbreak, several approaches have been given a try to quickly tackle this global calamity. One of the well-established strategies is the drug repurposing, which consists in finding new therapeutic uses for approved drugs. Following the same paradigm, we report in the present study, an investigation of the potential inhibitory activity of 5-FU and nineteen of its analogues against the SARS-CoV-2 main protease (3CLpro).
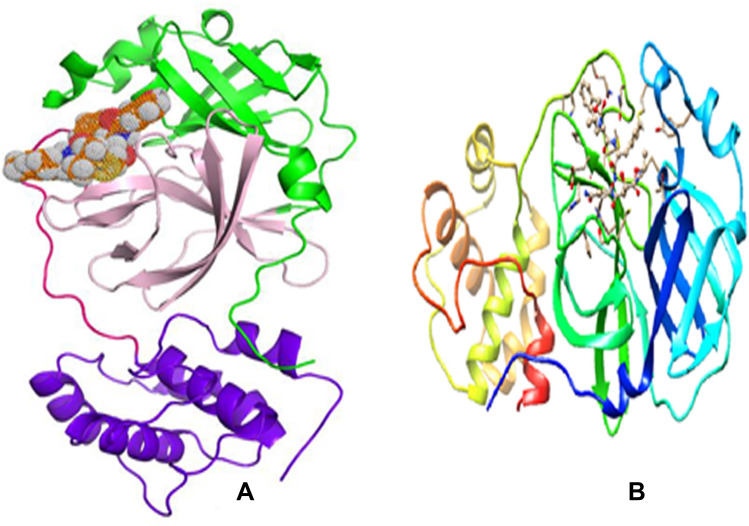
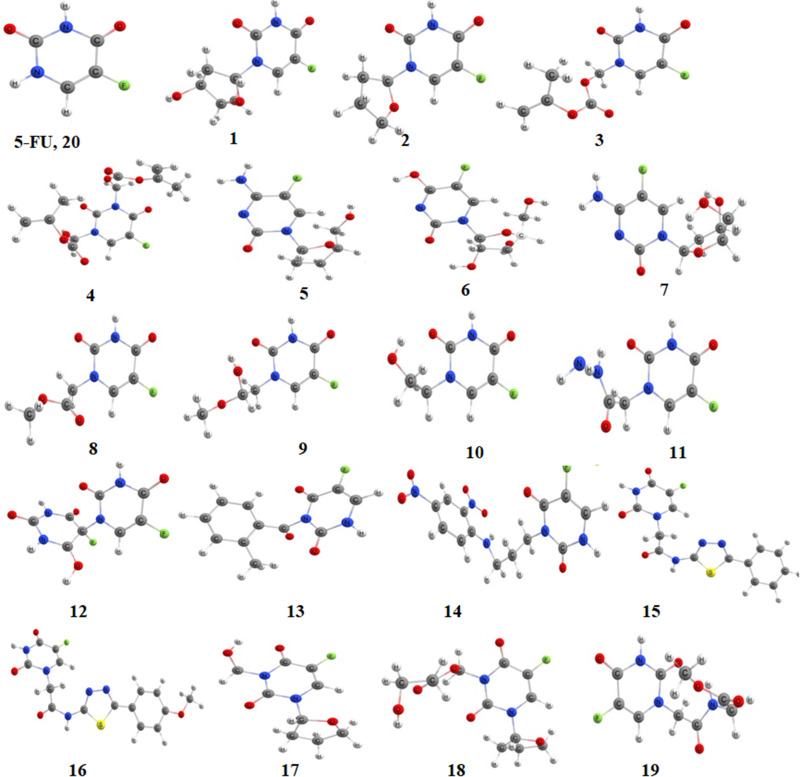
Material and methods: Molecular docking calculations were performed to investigate the binding affinity of the ligands within the active site of 3CLpro. The best binding candidates were further considered for molecular dynamics simulations for 100 ns to gain a time-resolved understanding of the behavior of the guest-host complexes. Furthermore, the profile of druggability of the best binding ligands was assessed based on ADMET predictions. Finally, their chemical reactivity was elucidated using different reactivity descriptors, namely the molecular electrostatic potential (MEP), Fukui functions and frontier molecular orbitals.
Results and discussion: From the calculations performed, four candidates (compounds 14, 15, 16 and 18) show promising results with respect to the binding affinity to the target protease, 3CLpro, the therapeutic profile of druggability and safety. These compounds are maintained inside the active site of 3CLpro thanks to a variety of noncovalent interactions, especially hydrogen bonds, involving important amino acids such as GLU166, HIS163, GLY143, ASN142, HIS172, CYS145. Molecular dynamics simulations suggest that the four ligands are well trapped within the active site of the protein over a time gap of 100 ns, ligand 18 being the most retained.
Conclusion: In line with the findings reported herein, we recommend that further in-vitro and in-vivo investigations are carried out to shed light on the possible mechanism of pharmacological action of the proposed ligands.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



