Rhegmatogenous Retinal Detachment in Waardenburg Syndrome: A Case Report.
Q3 Medicine
Korean Journal of Ophthalmology : KJO
Pub Date : 2022-10-01
Epub Date: 2022-08-19
DOI:10.3341/kjo.2022.0052
引用次数: 0
Abstract
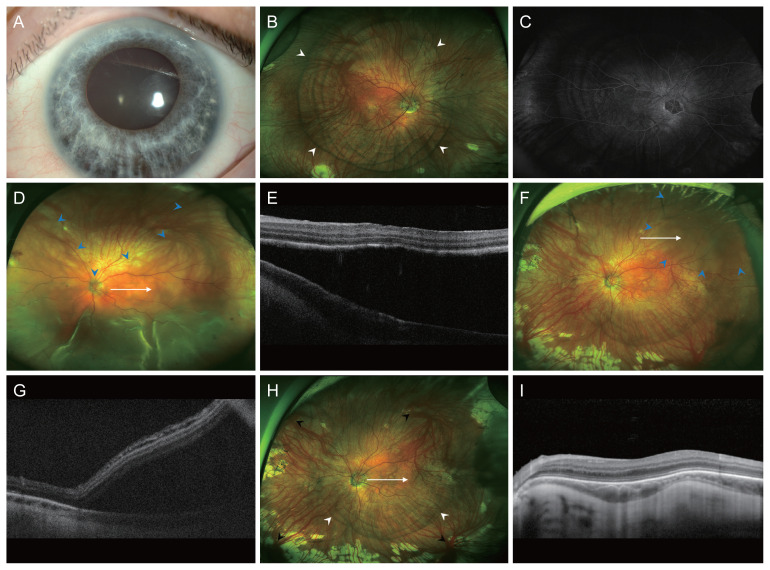
Dear Editor, Waardenburg syndrome (WS) is a rare auditory-pigmentary disorder and characterized by sensorineural hearing loss, pigmentary disturbances of the skin, hair, iris, and choroid [1]. WS is caused by mutations of several genes that affect the division and migration of neural crest cells (NCCs) during embryonic development. Rhegmatogenous retinal detachment (RRD) in WS has been rarely discussed in the literature. Herein, we report an extremely rare case of RRD in a patient with WS who was successfully treated with surgical management. Written informed consent was obtained from the patient A 24-year-old male patient diagnosed with WS was referred to Seoul National University Hospital with decreased visual acuity in his left eye (OS). On general examination, he had grey hair and skin hypopigmentation. He also had a cochlear implant because of congenital sensorineural hearing impairment. Genetic analysis revealed heterozygous pathogenic variant c.649_651del (p.Arg217del) of the MITF gene, confirming a diagnosis of WS type 2A. He had no history of ocular trauma or atopic dermatitis. His best-corrected visual acuity (BCVA) were 20 / 32 in the right eye (OD) and hand motion OS. The spherical equivalents were -6.88 diopters (D) OD and -4.63 D OS, and axial lengths were 26.89 mm OD and 23.44 mm OS. Slit-lamp examination showed heterochromia with blue iris (Fig. 1A). The right fundus revealed diffuse choroidal hypopigmentation and multiple choroidal pigmentation (Fig. 1B). On fluorescein angiography, blocked hypofluorescence corresponded to areas of choroidal hypopigmentation was noted (Fig. 1C). The left fundus demonstrates bullous, nearly total RRD (Fig. 1D). Because of poor mydriasis and choroidal hypopigmentation, no retinal break could be determined. Optical coherence tomography (OCT) showed foveal hypoplasia with macula-off RRD (Fig. 1E). The patient underwent a pars plana vitrectomy with silicone oil (SO) tamponade. During the surgery, retinal breaks were identified at 2 and 9 o’clock in the peripheral retina. One year after surgery, the BCVA was 20 / 40 OS, and the retina was completely f lat. Accordingly, SO was removed and cataract surgery was performed during vitrectomy. After SO removal, the retina remained f lat during the 23 months of follow-up. Two years after SO removal, he had sudden reduction of visual fields. His BCVA was 20 / 50 OS and fundus examination revealed recurred RRD in the superotemporal retina (Fig. 1F, 1G). He underwent repeated vitrectomy with gas tamponade (sulfur hexafluoride [SF6], 20%). Intraoperatively, a new retinal break was identified at 2 o’clock. After surgery, the retina was entirely flat. At the last visit, 6.5 years after the surgery, his BCVA was 20 / 40 OS, and the patient had no recurrence of RRD. Fundoscopy demonstrated diffuse choroidal hypopigmentation with multiple ring-shaped choroidal pigmentation in the left eye (Fig. 1H). OCT showed foveal hypoplasia and irregular bowing out of the choroidal-scleral interface (Fig. 1I). To the best of our knowledge, this is the first case of RRD in a patient with WS. WS is caused by variants in the EDN3, EDNRB, MITF, PAX3, SNAI2, and SOX10 genes which results in defective neural crest development and melanocyte migration [2]. During embryonic development, NCCs infiltrate the iris to provide its pigmentation and also differentiate into the pericytes of the choroid, as well as the sclera [3]. It is reasonable to speculate that choroidal concentric pigmentation, foveal hypoplasia, and irregular bowing out of the choroidal-scleral interface may be attributable to the defective melanocyte migration and developmental anomaly of embryogenesis derived from NCCs. However, it remains unclear whether the relationship between WS and RRD is causal or coincidental as of now. The diagnosis and treatment of RRD in WS are challenging. There is difficulty in finding the retinal breaks from hypopigmented choroid. The lack of pigment causing difficulties with efficient visualization of laser burn and Korean J Ophthalmol 2022;36(5):468-470 https://doi.org/10.3341/kjo.2022.0052
Waardenburg综合征致孔源性视网膜脱离1例。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


