Maximiliano L Cacicedo, Christine Weinl-Tenbruck, Daniel Frank, Sebastian Wirsching, Beate K Straub, Jana Hauke, Jürgen G Okun, Nigel Horscroft, Julia B Hennermann, Fred Zepp, Frédéric Chevessier-Tünnesen, Stephan Gehring
{"title":"mRNA-based therapy proves superior to the standard of care for treating hereditary tyrosinemia 1 in a mouse model.","authors":"Maximiliano L Cacicedo, Christine Weinl-Tenbruck, Daniel Frank, Sebastian Wirsching, Beate K Straub, Jana Hauke, Jürgen G Okun, Nigel Horscroft, Julia B Hennermann, Fred Zepp, Frédéric Chevessier-Tünnesen, Stephan Gehring","doi":"10.1016/j.omtm.2022.07.006","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary tyrosinemia type 1 is an inborn error of amino acid metabolism characterized by deficiency of fumarylacetoacetate hydrolase (FAH). Only limited treatment options (e.g., oral nitisinone) are available. Patients must adhere to a strict diet and face a life-long risk of complications, including liver cancer and progressive neurocognitive decline. There is a tremendous need for innovative therapies that standardize metabolite levels and promise normal development. Here, we describe an mRNA-based therapeutic approach that rescues <i>Fah</i>-deficient mice, a well-established tyrosinemia model. Repeated intravenous or intramuscular administration of lipid nanoparticle-formulated human <i>FAH</i> mRNA resulted in FAH protein synthesis in deficient mouse livers, stabilized body weight, normalized pathologic increases in metabolites after nitisinone withdrawal, and prevented early death. Dose reduction and extended injection intervals proved therapeutically effective. These results provide proof of concept for an mRNA-based therapeutic approach to treating hereditary tyrosinemia type 1 that is superior to the standard of care.</p>","PeriodicalId":517056,"journal":{"name":"Molecular Therapy. Methods & Clinical Development","volume":" ","pages":"294-308"},"PeriodicalIF":0.0000,"publicationDate":"2022-07-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/4c/12/main.PMC9357842.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Therapy. Methods & Clinical Development","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.omtm.2022.07.006","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/8 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 3
Abstract
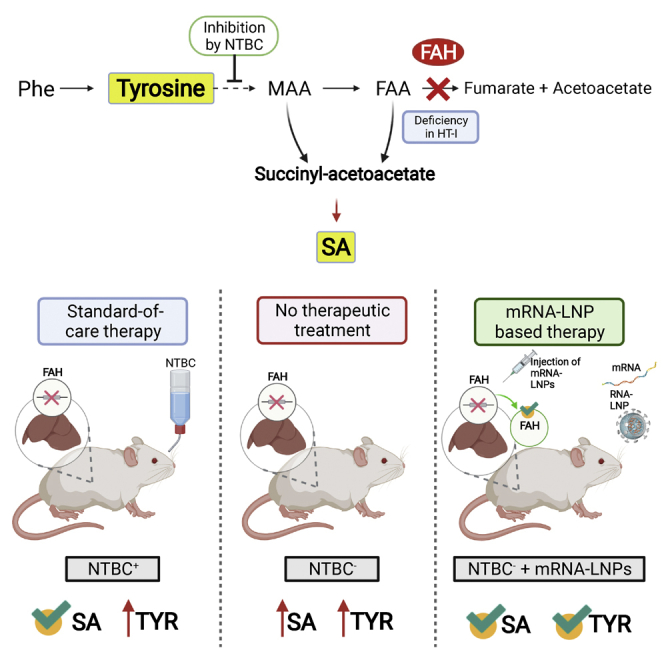
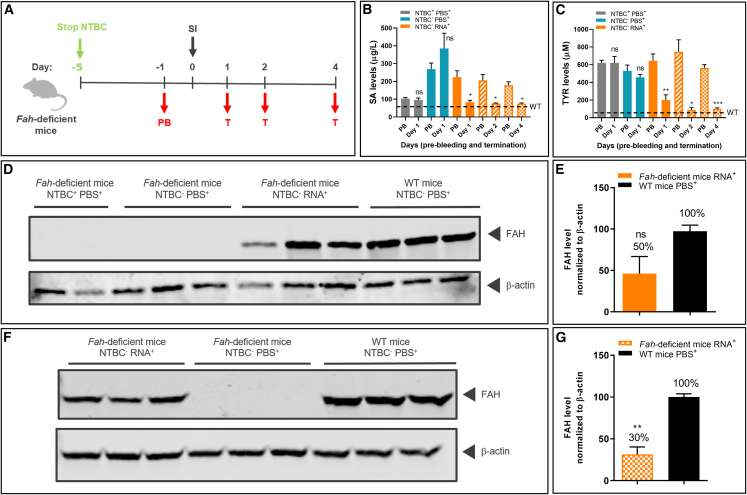
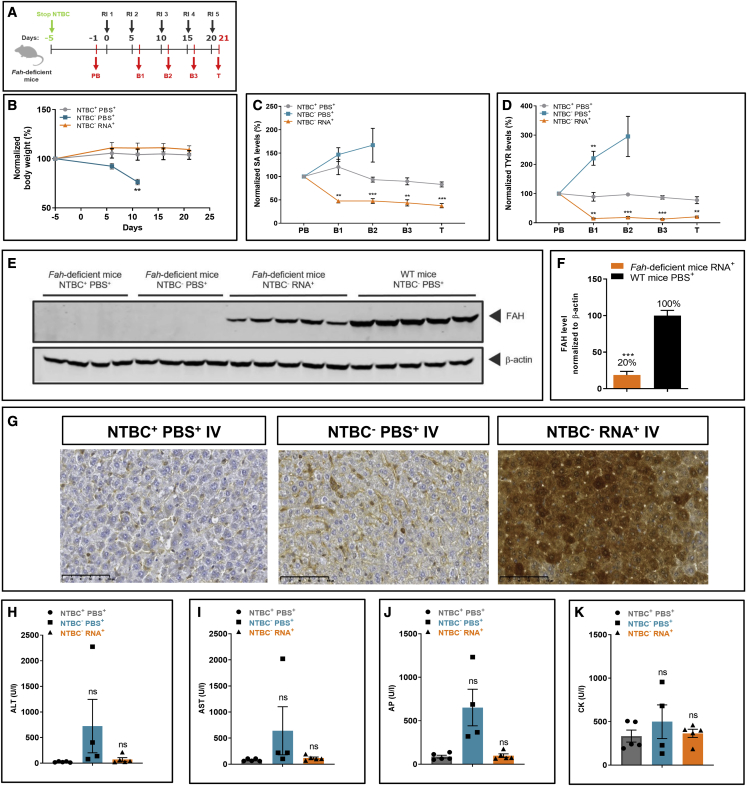
Hereditary tyrosinemia type 1 is an inborn error of amino acid metabolism characterized by deficiency of fumarylacetoacetate hydrolase (FAH). Only limited treatment options (e.g., oral nitisinone) are available. Patients must adhere to a strict diet and face a life-long risk of complications, including liver cancer and progressive neurocognitive decline. There is a tremendous need for innovative therapies that standardize metabolite levels and promise normal development. Here, we describe an mRNA-based therapeutic approach that rescues Fah-deficient mice, a well-established tyrosinemia model. Repeated intravenous or intramuscular administration of lipid nanoparticle-formulated human FAH mRNA resulted in FAH protein synthesis in deficient mouse livers, stabilized body weight, normalized pathologic increases in metabolites after nitisinone withdrawal, and prevented early death. Dose reduction and extended injection intervals proved therapeutically effective. These results provide proof of concept for an mRNA-based therapeutic approach to treating hereditary tyrosinemia type 1 that is superior to the standard of care.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


