{"title":"Binning long reads in metagenomics datasets using composition and coverage information.","authors":"Anuradha Wickramarachchi, Yu Lin","doi":"10.1186/s13015-022-00221-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Advancements in metagenomics sequencing allow the study of microbial communities directly from their environments. Metagenomics binning is a key step in the species characterisation of microbial communities. Next-generation sequencing reads are usually assembled into contigs for metagenomics binning mainly due to the limited information within short reads. Third-generation sequencing provides much longer reads that have lengths similar to the contigs assembled from short reads. However, existing contig-binning tools cannot be directly applied on long reads due to the absence of coverage information and the presence of high error rates. The few existing long-read binning tools either use only composition or use composition and coverage information separately. This may ignore bins that correspond to low-abundance species or erroneously split bins that correspond to species with non-uniform coverages. Here we present a reference-free binning approach, LRBinner, that combines composition and coverage information of complete long-read datasets. LRBinner also uses a distance-histogram-based clustering algorithm to extract clusters with varying sizes.</p><p><strong>Results: </strong>The experimental results on both simulated and real datasets show that LRBinner achieves the best binning accuracy in most cases while handling the complete datasets without any sampling. Moreover, we show that binning reads using LRBinner prior to assembly reduces computational resources required for assembly while attaining satisfactory assembly qualities.</p><p><strong>Conclusion: </strong>LRBinner shows that deep-learning techniques can be used for effective feature aggregation to support the metagenomics binning of long reads. Furthermore, accurate binning of long reads supports improvements in metagenomics assembly, especially in complex datasets. Binning also helps to reduce the resources required for assembly. Source code for LRBinner is freely available at https://github.com/anuradhawick/LRBinner.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":" ","pages":"14"},"PeriodicalIF":1.5000,"publicationDate":"2022-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9277797/pdf/","citationCount":"8","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-022-00221-z","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 8
Abstract
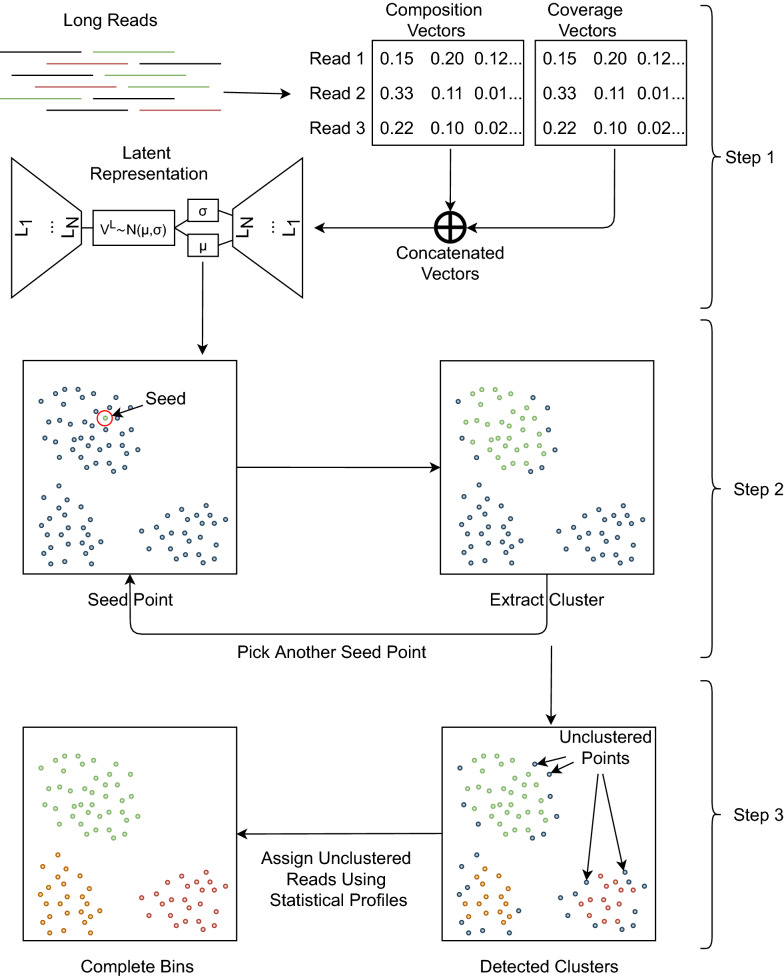
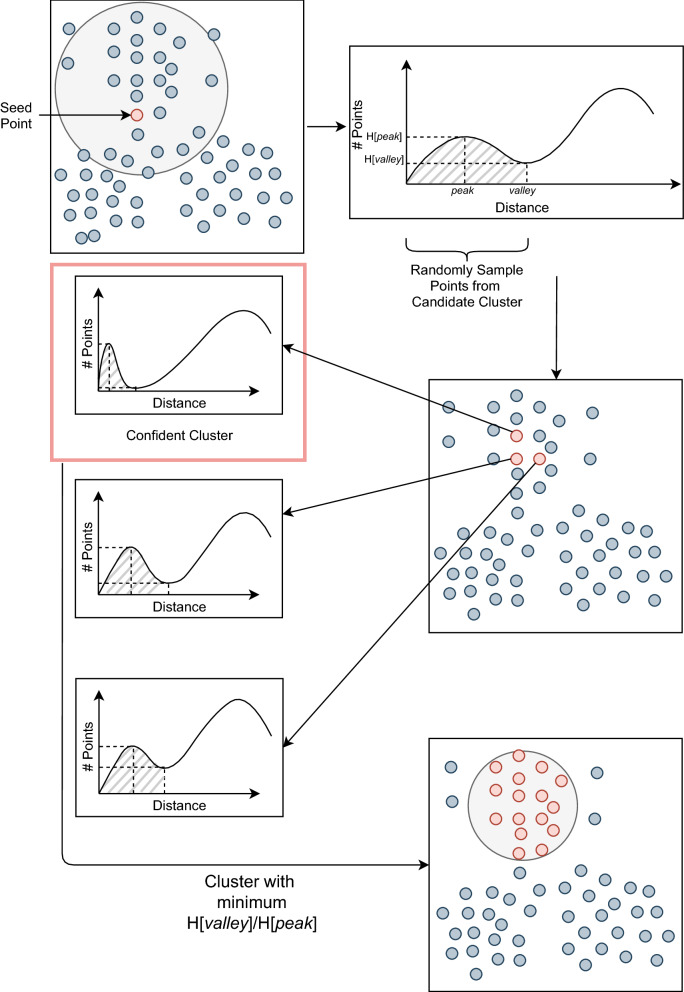
Background: Advancements in metagenomics sequencing allow the study of microbial communities directly from their environments. Metagenomics binning is a key step in the species characterisation of microbial communities. Next-generation sequencing reads are usually assembled into contigs for metagenomics binning mainly due to the limited information within short reads. Third-generation sequencing provides much longer reads that have lengths similar to the contigs assembled from short reads. However, existing contig-binning tools cannot be directly applied on long reads due to the absence of coverage information and the presence of high error rates. The few existing long-read binning tools either use only composition or use composition and coverage information separately. This may ignore bins that correspond to low-abundance species or erroneously split bins that correspond to species with non-uniform coverages. Here we present a reference-free binning approach, LRBinner, that combines composition and coverage information of complete long-read datasets. LRBinner also uses a distance-histogram-based clustering algorithm to extract clusters with varying sizes.
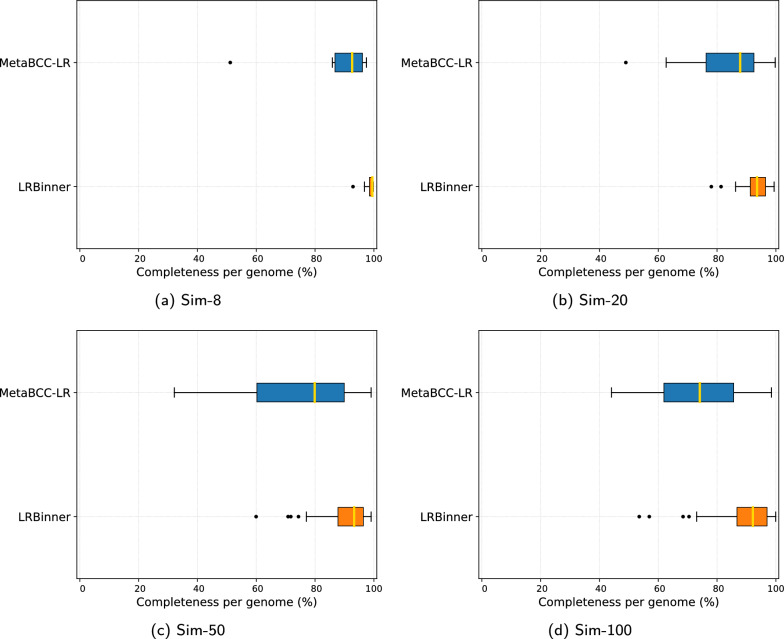
Results: The experimental results on both simulated and real datasets show that LRBinner achieves the best binning accuracy in most cases while handling the complete datasets without any sampling. Moreover, we show that binning reads using LRBinner prior to assembly reduces computational resources required for assembly while attaining satisfactory assembly qualities.
Conclusion: LRBinner shows that deep-learning techniques can be used for effective feature aggregation to support the metagenomics binning of long reads. Furthermore, accurate binning of long reads supports improvements in metagenomics assembly, especially in complex datasets. Binning also helps to reduce the resources required for assembly. Source code for LRBinner is freely available at https://github.com/anuradhawick/LRBinner.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


