Srinath Pashikanti, Daniel J. Foster, Yugesh Kharel, Anne M. Brown, David R. Bevan, Kevin R. Lynch and Webster L. Santos*,
{"title":"Sphingosine Kinase 2 Inhibitors: Rigid Aliphatic Tail Derivatives Deliver Potent and Selective Analogues","authors":"Srinath Pashikanti, Daniel J. Foster, Yugesh Kharel, Anne M. Brown, David R. Bevan, Kevin R. Lynch and Webster L. Santos*, ","doi":"10.1021/acsbiomedchemau.2c00017","DOIUrl":null,"url":null,"abstract":"<p >Sphingosine 1-phosphate (S1P) is a pleiotropic signaling molecule that interacts with five native G-protein coupled receptors (S1P1–5) to regulate cell growth, survival, and proliferation. S1P has been implicated in a variety of pathologies including cancer, kidney fibrosis, and multiple sclerosis. As key mediators in the synthesis of S1P, sphingosine kinase (SphK) isoforms 1 and 2 have attracted attention as viable targets for pharmacologic intervention. In this report, we describe the design, synthesis, and biological evaluation of sphingosine kinase 2 (SphK2) inhibitors with a focus on systematically introducing rigid structures in the aliphatic lipid tail present in existing SphK2 inhibitors. Experimental as well as molecular modeling studies suggest that conformationally restricted “lipophilic tail” analogues bearing a bulky terminal moiety or an internal phenyl ring are useful to complement the “J”-shaped sphingosine binding pocket of SphK2. We identified <b>14c</b> (SLP9101555) as a potent SphK2 inhibitor (<i>K</i><sub>i</sub> = 90 nM) with 200-fold selectivity over SphK1. Molecular docking studies indicated key interactions: the cyclohexyl ring binding in the cleft deep in the pocket, a trifluoromethyl group fitting in a small side cavity, and a hydrogen bond between the guanidino group and Asp308 (amino acid numbering refers to human SphK2 (isoform c) orthologue). <i>In vitro</i> studies using U937 human histiocytic lymphoma cells showed marked decreases in extracellular S1P levels in response to our SphK2 inhibitors. Administration of <b>14c</b> (dose: 5 mg/kg) to mice resulted in a sustained increase of circulating S1P levels, suggesting target engagement.</p>","PeriodicalId":29802,"journal":{"name":"ACS Bio & Med Chem Au","volume":"2 5","pages":"469–489"},"PeriodicalIF":3.8000,"publicationDate":"2022-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c7/42/bg2c00017.PMC9585524.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Bio & Med Chem Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
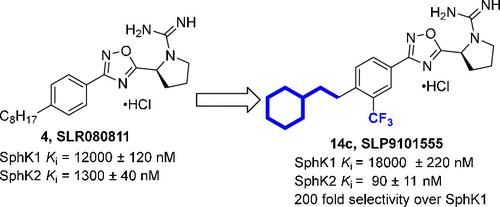
Sphingosine 1-phosphate (S1P) is a pleiotropic signaling molecule that interacts with five native G-protein coupled receptors (S1P1–5) to regulate cell growth, survival, and proliferation. S1P has been implicated in a variety of pathologies including cancer, kidney fibrosis, and multiple sclerosis. As key mediators in the synthesis of S1P, sphingosine kinase (SphK) isoforms 1 and 2 have attracted attention as viable targets for pharmacologic intervention. In this report, we describe the design, synthesis, and biological evaluation of sphingosine kinase 2 (SphK2) inhibitors with a focus on systematically introducing rigid structures in the aliphatic lipid tail present in existing SphK2 inhibitors. Experimental as well as molecular modeling studies suggest that conformationally restricted “lipophilic tail” analogues bearing a bulky terminal moiety or an internal phenyl ring are useful to complement the “J”-shaped sphingosine binding pocket of SphK2. We identified 14c (SLP9101555) as a potent SphK2 inhibitor (Ki = 90 nM) with 200-fold selectivity over SphK1. Molecular docking studies indicated key interactions: the cyclohexyl ring binding in the cleft deep in the pocket, a trifluoromethyl group fitting in a small side cavity, and a hydrogen bond between the guanidino group and Asp308 (amino acid numbering refers to human SphK2 (isoform c) orthologue). In vitro studies using U937 human histiocytic lymphoma cells showed marked decreases in extracellular S1P levels in response to our SphK2 inhibitors. Administration of 14c (dose: 5 mg/kg) to mice resulted in a sustained increase of circulating S1P levels, suggesting target engagement.
期刊介绍:
ACS Bio & Med Chem Au is a broad scope open access journal which publishes short letters comprehensive articles reviews and perspectives in all aspects of biological and medicinal chemistry. Studies providing fundamental insights or describing novel syntheses as well as clinical or other applications-based work are welcomed.This broad scope includes experimental and theoretical studies on the chemical physical mechanistic and/or structural basis of biological or cell function in all domains of life. It encompasses the fields of chemical biology synthetic biology disease biology cell biology agriculture and food natural products research nucleic acid biology neuroscience structural biology and biophysics.The journal publishes studies that pertain to a broad range of medicinal chemistry including compound design and optimization biological evaluation molecular mechanistic understanding of drug delivery and drug delivery systems imaging agents and pharmacology and translational science of both small and large bioactive molecules. Novel computational cheminformatics and structural studies for the identification (or structure-activity relationship analysis) of bioactive molecules ligands and their targets are also welcome. The journal will consider computational studies applying established computational methods but only in combination with novel and original experimental data (e.g. in cases where new compounds have been designed and tested).Also included in the scope of the journal are articles relating to infectious diseases research on pathogens host-pathogen interactions therapeutics diagnostics vaccines drug-delivery systems and other biomedical technology development pertaining to infectious diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


