Wesley Schaal, Adam Ameur, Ulla Olsson-Strömberg, Monica Hermanson, Lucia Cavelier, Ola Spjuth
{"title":"Migrating to Long-Read Sequencing for Clinical Routine <i>BCR-ABL1</i> TKI Resistance Mutation Screening.","authors":"Wesley Schaal, Adam Ameur, Ulla Olsson-Strömberg, Monica Hermanson, Lucia Cavelier, Ola Spjuth","doi":"10.1177/11769351221110872","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>The aim of this project was to implement long-read sequencing for BCR-ABL1 TKI resistance mutation screening in a clinical setting for patients undergoing treatment for chronic myeloid leukemia.</p><p><strong>Materials and methods: </strong>Processes were established for registering and transferring samples from the clinic to an academic sequencing facility for long-read sequencing. An automated analysis pipeline for detecting mutations was established, and an information system was implemented comprising features for data management, analysis and visualization. Clinical validation was performed by identifying BCR-ABL1 TKI resistance mutations by Sanger and long-read sequencing in parallel. The developed software is available as open source via GitHub at https://github.com/pharmbio/clamp.</p><p><strong>Results: </strong>The information system enabled traceable transfer of samples from the clinic to the sequencing facility, robust and automated analysis of the long-read sequence data, and communication of results from sequence analysis in a reporting format that could be easily interpreted and acted upon by clinical experts. In a validation study, all 17 resistance mutations found by Sanger sequencing were also detected by long-read sequencing. An additional 16 mutations were found only by long-read sequencing, all of them with frequencies below the limit of detection for Sanger sequencing. The clonal distributions of co-existing mutations were automatically resolved through the long-read data analysis. After the implementation and validation, the clinical laboratory switched their routine protocol from using Sanger to long-read sequencing for this application.</p><p><strong>Conclusions: </strong>Long-read sequencing delivers results with higher sensitivity compared to Sanger sequencing and enables earlier detection of emerging TKI resistance mutations. The developed processes, analysis workflow, and software components lower barriers for adoption and could be extended to other applications.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":" ","pages":"11769351221110872"},"PeriodicalIF":2.5000,"publicationDate":"2022-07-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9290162/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351221110872","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Objective: The aim of this project was to implement long-read sequencing for BCR-ABL1 TKI resistance mutation screening in a clinical setting for patients undergoing treatment for chronic myeloid leukemia.
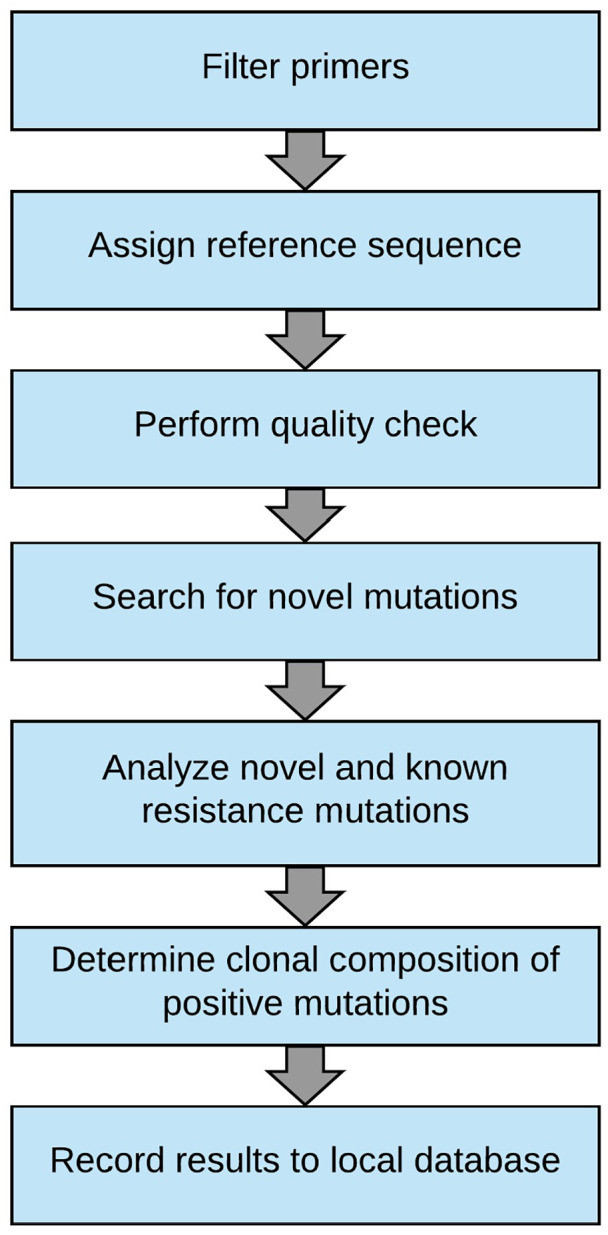
Materials and methods: Processes were established for registering and transferring samples from the clinic to an academic sequencing facility for long-read sequencing. An automated analysis pipeline for detecting mutations was established, and an information system was implemented comprising features for data management, analysis and visualization. Clinical validation was performed by identifying BCR-ABL1 TKI resistance mutations by Sanger and long-read sequencing in parallel. The developed software is available as open source via GitHub at https://github.com/pharmbio/clamp.
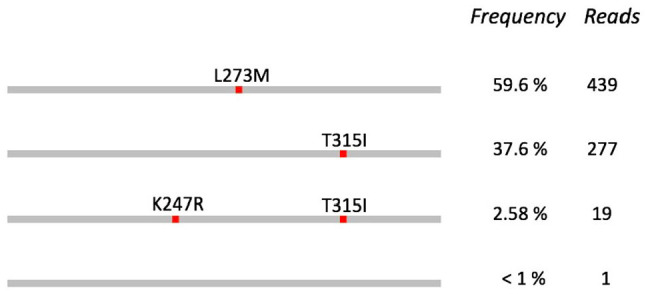
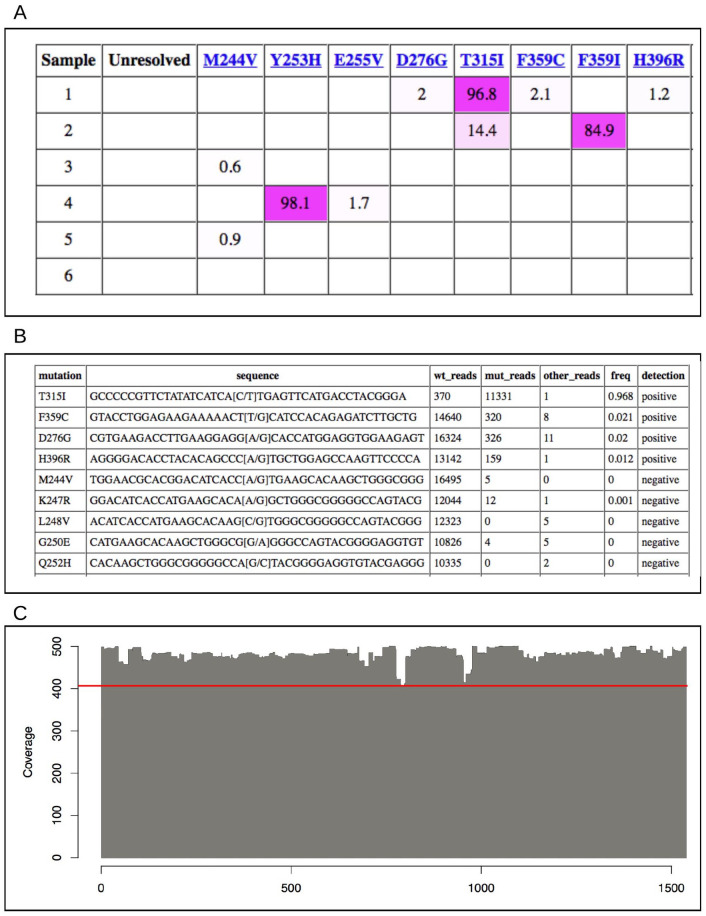
Results: The information system enabled traceable transfer of samples from the clinic to the sequencing facility, robust and automated analysis of the long-read sequence data, and communication of results from sequence analysis in a reporting format that could be easily interpreted and acted upon by clinical experts. In a validation study, all 17 resistance mutations found by Sanger sequencing were also detected by long-read sequencing. An additional 16 mutations were found only by long-read sequencing, all of them with frequencies below the limit of detection for Sanger sequencing. The clonal distributions of co-existing mutations were automatically resolved through the long-read data analysis. After the implementation and validation, the clinical laboratory switched their routine protocol from using Sanger to long-read sequencing for this application.
Conclusions: Long-read sequencing delivers results with higher sensitivity compared to Sanger sequencing and enables earlier detection of emerging TKI resistance mutations. The developed processes, analysis workflow, and software components lower barriers for adoption and could be extended to other applications.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


