{"title":"Autosomal dominant Ullrich congenital muscular dystrophy due to a <i>de novo</i> mutation in <i>COL6A3</i> gene. A case report.","authors":"Esther Picillo, Annalaura Torella, Luigia Passamano, Vincenzo Nigro, Luisa Politano","doi":"10.36185/2532-1900-073","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in the genes encoding collagen VI cause Bethlem myopathy (MIM 158810), Ullrich congenital muscular dystrophy (MIM 254090), and myosclerosis myopathy (MIM #255600). BM is a dominantly inherited disorder, characterised by proximal muscle weakness and joint contractures mainly involving the elbows, ankles, and fingers, which usually follows a relatively mild course. By contrast, UCMD is a severe muscular dystrophy characterized by early onset, rapidly progressive muscle wasting and weakness, proximal joint contractures and distal joint hyperlaxity. Rapid progression usually leads to early death due to respiratory failure. UCMD is usually inherited as an autosomal recessive trait though dominant <i>de novo</i> heterozygous variants have recently been reported. We describe a further patient with UCMD classical presentation who showed, at the NGS analysis, the <i>de novo</i> variant c.6210+1G > A in the intron 16 of the gene <i>COL6A3</i>, known in the literature as pathogenic (VCV0000949S6.5).</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"41 2","pages":"95-98"},"PeriodicalIF":0.0000,"publicationDate":"2022-06-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/aa/54/am-2022-02-95.PMC9237747.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-073","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 1
Abstract
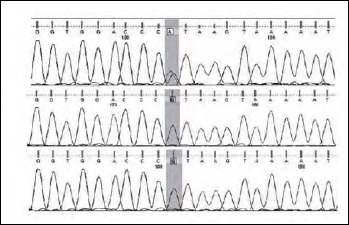
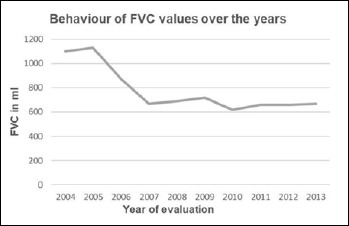
Mutations in the genes encoding collagen VI cause Bethlem myopathy (MIM 158810), Ullrich congenital muscular dystrophy (MIM 254090), and myosclerosis myopathy (MIM #255600). BM is a dominantly inherited disorder, characterised by proximal muscle weakness and joint contractures mainly involving the elbows, ankles, and fingers, which usually follows a relatively mild course. By contrast, UCMD is a severe muscular dystrophy characterized by early onset, rapidly progressive muscle wasting and weakness, proximal joint contractures and distal joint hyperlaxity. Rapid progression usually leads to early death due to respiratory failure. UCMD is usually inherited as an autosomal recessive trait though dominant de novo heterozygous variants have recently been reported. We describe a further patient with UCMD classical presentation who showed, at the NGS analysis, the de novo variant c.6210+1G > A in the intron 16 of the gene COL6A3, known in the literature as pathogenic (VCV0000949S6.5).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


