Disease Course and Outcomes in Patients With the Limited Form of Neuromyelitis Optica Spectrum Disorders and Negative AQP4-IgG Serology at Disease Onset: A Prospective Cohort Study.
{"title":"Disease Course and Outcomes in Patients With the Limited Form of Neuromyelitis Optica Spectrum Disorders and Negative AQP4-IgG Serology at Disease Onset: A Prospective Cohort Study.","authors":"Xiaodong Chen, Jing Zhou, Rui Li, Bingjun Zhang, Yuge Wang, Xiaonan Zhong, Yaqing Shu, Yanyu Chang, Wei Qiu","doi":"10.3988/jcn.2022.18.4.453","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and purpose: </strong>Patients presenting with clinical characteristics that are strongly suggestive of neuromyelitis optica spectrum disorders (NMOSD) have a high risk of developing definite NMOSD in the future. Little is known about the clinical course, treatment, and prognosis of these patients with likely NMOSD at disease onset.</p><p><strong>Methods: </strong>This study prospectively recruited and visited 24 patients with the limited form of NMOSD (LF-NMOSD) at disease onset from November 2012 to June 2021. Their demographics, clinical course, longitudinal aquaporin-4 immunoglobulin G (AQP4-IgG) serology, MRI, therapeutic management, and outcome data were collected and analyzed.</p><p><strong>Results: </strong>The onset age of the cohort was 38.1±12.0 years (mean±standard deviation). The median disease duration was 73.5 months (interquartile range=44.3-117.0 months), and the follow-up period was 54.2±23.8 months. At the end of the last visit, the final diagnosis was categorized into AQP4-IgG-seronegative NMOSD (<i>n</i>=16, 66.7%), AQP4-IgG-seropositive NMOSD (<i>n</i>=7, 29.2%), or multiple sclerosis (<i>n</i>=1, 4.2%). Seven of the 24 patients (29.2%) experienced conversion to AQP4-IgG seropositivity, and the interval from onset to this serological conversion was 37.9±21.9 months. Isolated/mixed area postrema syndrome (APS) was the predominant onset phenotype (37.5%). The patients with isolated/mixed APS onset showed a predilection for conversion to AQP4-IgG seropositivity. All patients experienced a multiphasic disease course, with immunosuppressive therapy reducing the incidence rates of clinical relapse and residual functional disability.</p><p><strong>Conclusions: </strong>Definite NMOSD may be preceded by LF-NMOSD, particularly isolated/mixed APS. Intensive long-term follow-up and attack-prevention immunotherapeutic management is recommended in patients with LF-NMOSD.</p>","PeriodicalId":324902,"journal":{"name":"Journal of Clinical Neurology (Seoul, Korea)","volume":" ","pages":"453-462"},"PeriodicalIF":0.0000,"publicationDate":"2022-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/5c/71/jcn-18-453.PMC9262456.pdf","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Neurology (Seoul, Korea)","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3988/jcn.2022.18.4.453","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
Abstract
Background and purpose: Patients presenting with clinical characteristics that are strongly suggestive of neuromyelitis optica spectrum disorders (NMOSD) have a high risk of developing definite NMOSD in the future. Little is known about the clinical course, treatment, and prognosis of these patients with likely NMOSD at disease onset.
Methods: This study prospectively recruited and visited 24 patients with the limited form of NMOSD (LF-NMOSD) at disease onset from November 2012 to June 2021. Their demographics, clinical course, longitudinal aquaporin-4 immunoglobulin G (AQP4-IgG) serology, MRI, therapeutic management, and outcome data were collected and analyzed.
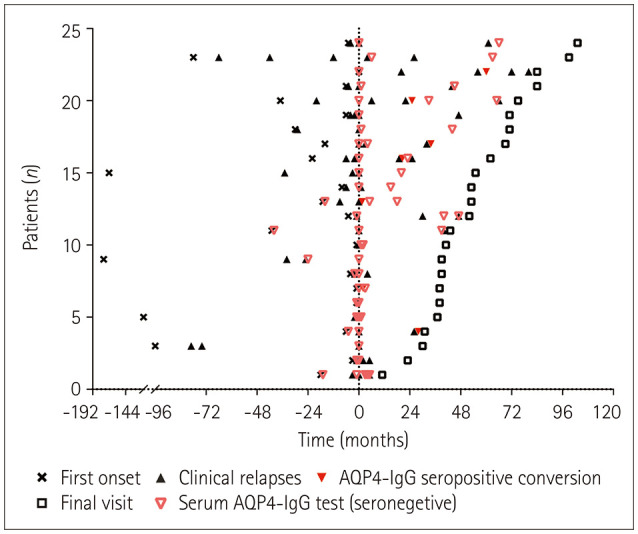
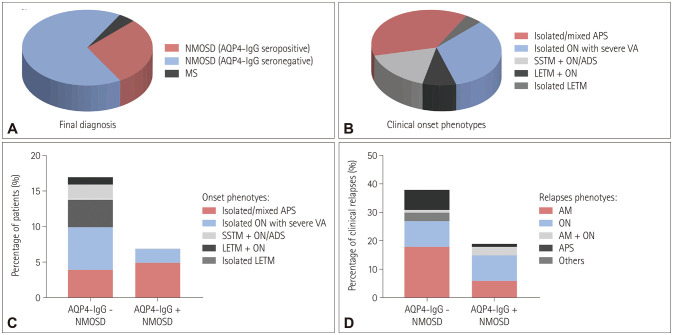
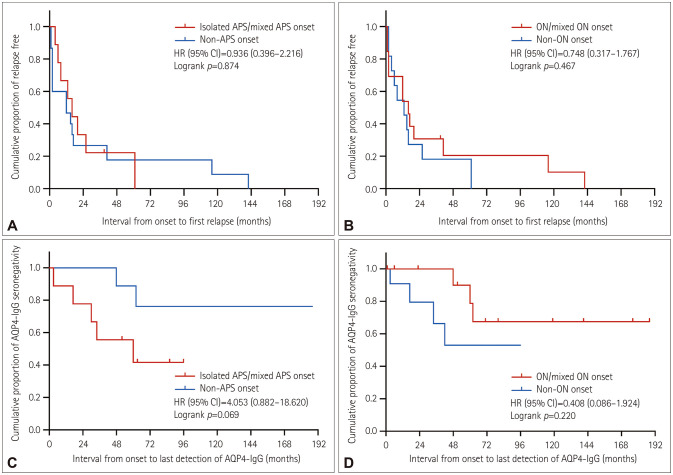
Results: The onset age of the cohort was 38.1±12.0 years (mean±standard deviation). The median disease duration was 73.5 months (interquartile range=44.3-117.0 months), and the follow-up period was 54.2±23.8 months. At the end of the last visit, the final diagnosis was categorized into AQP4-IgG-seronegative NMOSD (n=16, 66.7%), AQP4-IgG-seropositive NMOSD (n=7, 29.2%), or multiple sclerosis (n=1, 4.2%). Seven of the 24 patients (29.2%) experienced conversion to AQP4-IgG seropositivity, and the interval from onset to this serological conversion was 37.9±21.9 months. Isolated/mixed area postrema syndrome (APS) was the predominant onset phenotype (37.5%). The patients with isolated/mixed APS onset showed a predilection for conversion to AQP4-IgG seropositivity. All patients experienced a multiphasic disease course, with immunosuppressive therapy reducing the incidence rates of clinical relapse and residual functional disability.
Conclusions: Definite NMOSD may be preceded by LF-NMOSD, particularly isolated/mixed APS. Intensive long-term follow-up and attack-prevention immunotherapeutic management is recommended in patients with LF-NMOSD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


