Nan Geng, Guojin Sun, Wen-Jia Liu, Bin-Cheng Gao, Cong Sun, Cundong Xu, Ertian Hua, Lin Xu
{"title":"Distribution, Phylogeny and Evolution of Clinical and Environmental <i>Vibrio vulnificus</i> Antibiotic-Resistant Genes.","authors":"Nan Geng, Guojin Sun, Wen-Jia Liu, Bin-Cheng Gao, Cong Sun, Cundong Xu, Ertian Hua, Lin Xu","doi":"10.1177/11769343221134400","DOIUrl":null,"url":null,"abstract":"<p><p><i>Vibrio vulnificus</i> is an emergent marine pathogen and is the cause of a deadly septicemia. However, the evolution mechanism of antibiotic-resistant genes (ARGs) is still unclear. Twenty-two high-quality complete genomes of <i>V. vulnificus</i> were obtained and grouped into 16 clinical isolates and 6 environmental isolates. Genomic annotations found 23 ARG orthologous genes, among which 14 ARGs were shared by <i>V. vulnificus</i> and other <i>Vibrio</i> members. Furthermore, those ARGs were located in their chromosomes, rather than in the plasmids. Phylogenomic reconstruction based on single-copy orthologous protein sequences and ARG protein sequences revealed that clinical and environmental <i>V. vulnificus</i> isolates were in a scattered distribution. The calculation of non-synonymous and synonymous substitutions indicated that most of ARGs evolved under purifying selection with the <i>Ka</i>/<i>Ks</i> ratios lower than one, while <i>h-ns, rsmA</i>, and <i>soxR</i> in several clinical isolates evolved under the positive selection with <i>Ka</i>/<i>Ks</i> ratios >1. Our result indicated that <i>V. vulnificus</i> antibiotic-resistant armory was not only confined to clinical isolates, but to environmental ones as well and clinical isolates inclined to accumulate beneficial non-synonymous substitutions that could be retained to improve competitiveness.</p>","PeriodicalId":136690,"journal":{"name":"Evolutionary Bioinformatics Online","volume":" ","pages":"11769343221134400"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e0/04/10.1177_11769343221134400.PMC9669696.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics Online","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/11769343221134400","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
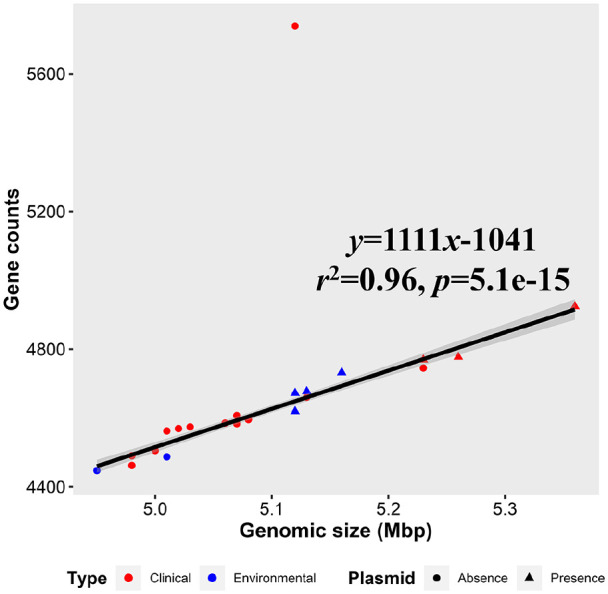
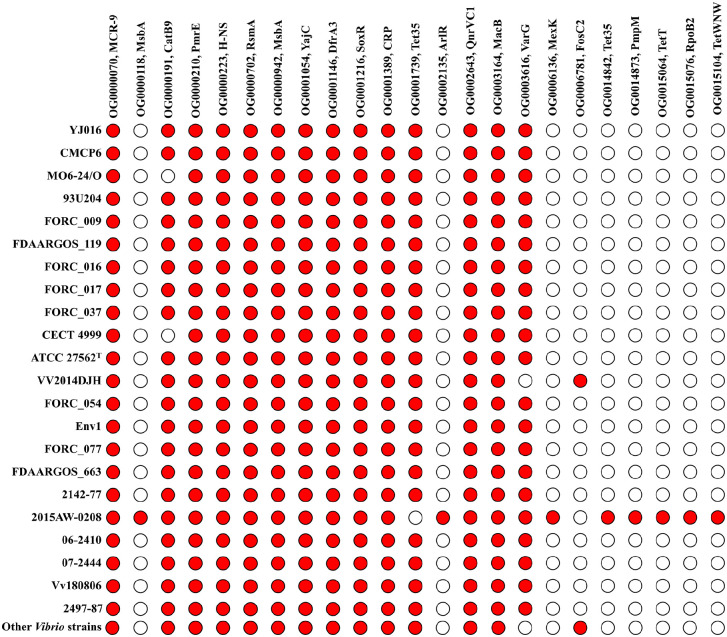
Vibrio vulnificus is an emergent marine pathogen and is the cause of a deadly septicemia. However, the evolution mechanism of antibiotic-resistant genes (ARGs) is still unclear. Twenty-two high-quality complete genomes of V. vulnificus were obtained and grouped into 16 clinical isolates and 6 environmental isolates. Genomic annotations found 23 ARG orthologous genes, among which 14 ARGs were shared by V. vulnificus and other Vibrio members. Furthermore, those ARGs were located in their chromosomes, rather than in the plasmids. Phylogenomic reconstruction based on single-copy orthologous protein sequences and ARG protein sequences revealed that clinical and environmental V. vulnificus isolates were in a scattered distribution. The calculation of non-synonymous and synonymous substitutions indicated that most of ARGs evolved under purifying selection with the Ka/Ks ratios lower than one, while h-ns, rsmA, and soxR in several clinical isolates evolved under the positive selection with Ka/Ks ratios >1. Our result indicated that V. vulnificus antibiotic-resistant armory was not only confined to clinical isolates, but to environmental ones as well and clinical isolates inclined to accumulate beneficial non-synonymous substitutions that could be retained to improve competitiveness.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


