{"title":"Congenital myopathy associated with a novel mutation in <i>MEGF10</i> gene, myofibrillar alteration and progressive course.","authors":"Carolina Croci, Monica Traverso, Serena Baratto, Michele Iacomino, Marina Pedemonte, Francesco Caroli, Marcello Scala, Claudio Bruno, Chiara Fiorillo","doi":"10.36185/2532-1900-076","DOIUrl":null,"url":null,"abstract":"<p><p>Early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the <i>MEGF10</i> gene (OMIM #614399). Phenotypic spectrum of EMARDD is variable, ranging from severe infantile forms in which patients are ventilator-dependent and die in childhood, to milder chronic disorders with a more favorable course (mild variant, mvEMARDD). Here we describe a 22 years old boy, offspring of consanguineous parents, presenting a congenital myopathic phenotype since infancy with elbow contractures and scoliosis. The patient developed a slowly progressive muscle weakness with impaired walking, rhinolalia, dysphagia, and respiratory involvement, which required noninvasive ventilation therapy since the age of 16 years. First muscle biopsy revealed unspecific muscle damage, with fiber size variation, internal nuclei and fibrosis. Myofibrillar alterations were noted at a second muscle biopsy including whorled fibres, cytoplasmic inclusion and minicores. Exome sequencing identified a homozygous mutation in <i>MEGF10</i> gene, c.2096G > C (p.Cys699Ser), inherited by both parents. This variant, not reported in public databases of mutations, is expected to alter the structure of the protein and is therefore predicted to be probably damaging according to ACMG classification. In conclusion, we found a new likely pathogenic mutation in <i>MEGF10</i>, which is responsible for a progressive form of mvEMARDD with myofibrillar alterations at muscle biopsy. Interestingly, the presence of <i>MEGF10</i> mutations has not been reported in Italian population. Early diagnosis of MEGF10 myopathy is essential in light of recent results from in vivo testing demonstrating a potential therapeutic effect of SSRIs compounds.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"41 3","pages":"111-116"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/34/97/am-2022-03-111.PMC9628799.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-076","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
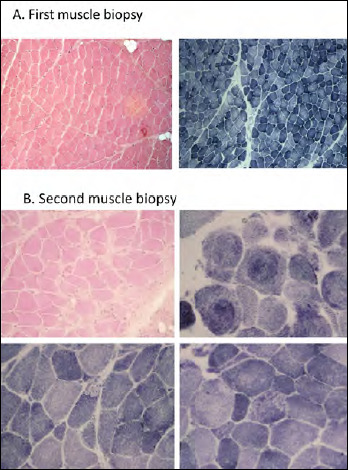
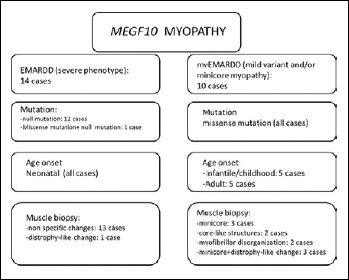
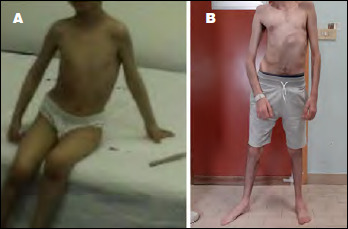
Early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the MEGF10 gene (OMIM #614399). Phenotypic spectrum of EMARDD is variable, ranging from severe infantile forms in which patients are ventilator-dependent and die in childhood, to milder chronic disorders with a more favorable course (mild variant, mvEMARDD). Here we describe a 22 years old boy, offspring of consanguineous parents, presenting a congenital myopathic phenotype since infancy with elbow contractures and scoliosis. The patient developed a slowly progressive muscle weakness with impaired walking, rhinolalia, dysphagia, and respiratory involvement, which required noninvasive ventilation therapy since the age of 16 years. First muscle biopsy revealed unspecific muscle damage, with fiber size variation, internal nuclei and fibrosis. Myofibrillar alterations were noted at a second muscle biopsy including whorled fibres, cytoplasmic inclusion and minicores. Exome sequencing identified a homozygous mutation in MEGF10 gene, c.2096G > C (p.Cys699Ser), inherited by both parents. This variant, not reported in public databases of mutations, is expected to alter the structure of the protein and is therefore predicted to be probably damaging according to ACMG classification. In conclusion, we found a new likely pathogenic mutation in MEGF10, which is responsible for a progressive form of mvEMARDD with myofibrillar alterations at muscle biopsy. Interestingly, the presence of MEGF10 mutations has not been reported in Italian population. Early diagnosis of MEGF10 myopathy is essential in light of recent results from in vivo testing demonstrating a potential therapeutic effect of SSRIs compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


