Whole-genome sequencing analysis of an atypical teratoid/rhabdoid tumor in a patient with Phelan-McDermid syndrome: a case report and systematic review.
{"title":"Whole-genome sequencing analysis of an atypical teratoid/rhabdoid tumor in a patient with Phelan-McDermid syndrome: a case report and systematic review.","authors":"Haruki Yamashita, Yoshiki Arakawa, Yukinori Terada, Yasuhide Takeuchi, Yohei Mineharu, Sosuke Sumiyoshi, Shinya Tokunaga, Kohei Nakajima, Naoko Kawabata, Kuniaki Tanaka, Masahiro Tanji, Katsutsugu Umeda, Sachiko Minamiguchi, Seishi Ogawa, Hironori Haga, Junko Takita, Susumu Miyamoto","doi":"10.1007/s10014-022-00440-7","DOIUrl":null,"url":null,"abstract":"<p><p>Atypical teratoid/rhabdoid tumor (AT/RT) is a rare pediatric brain tumor with abnormalities in SMARCB1 located in 22q11.2. We report a case of AT/RT associated with Phelan-McDermid syndrome (PMS) characterized by congenital developmental disorder, mental retardation, and ring chromosome 22 with 22q13.3-qter depletion, for which we performed whole-genome sequencing (WGS). A 4-year-old girl with a developmental disability was referred to our hospital due to dysphoria. Brain magnetic resonance imaging showed a 5-cm well-demarcated mass that extended bilaterally in the frontal lobes. G-banding was performed preoperatively due to a history of developmental retardation. Ring chromosome 22 and deletion of 22q13.3-qter were observed, and she was diagnosed with PMS. She underwent gross total resection of the tumor, and the pathological diagnosis was AT/RT. WGS showed somatic SMARCB1 mutation (p.R201X) and somatic loss of the entire chromosome 22 in the tumor, but not in the blood sample. WGS confirmed previously unreported BRCA2 mutations, 6q loss, and 14q acquisition during tumor progression, but no other significant findings associated with tumor progression. The present case is discussed with reference to a systematic review of previous reports of AT/RT associated with PMS. PMS patients with ring chromosome 22 should be carefully followed up for AT/RT occurrence.</p>","PeriodicalId":9226,"journal":{"name":"Brain Tumor Pathology","volume":" ","pages":"232-239"},"PeriodicalIF":3.0000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Tumor Pathology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10014-022-00440-7","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/24 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
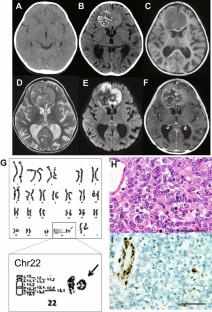
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare pediatric brain tumor with abnormalities in SMARCB1 located in 22q11.2. We report a case of AT/RT associated with Phelan-McDermid syndrome (PMS) characterized by congenital developmental disorder, mental retardation, and ring chromosome 22 with 22q13.3-qter depletion, for which we performed whole-genome sequencing (WGS). A 4-year-old girl with a developmental disability was referred to our hospital due to dysphoria. Brain magnetic resonance imaging showed a 5-cm well-demarcated mass that extended bilaterally in the frontal lobes. G-banding was performed preoperatively due to a history of developmental retardation. Ring chromosome 22 and deletion of 22q13.3-qter were observed, and she was diagnosed with PMS. She underwent gross total resection of the tumor, and the pathological diagnosis was AT/RT. WGS showed somatic SMARCB1 mutation (p.R201X) and somatic loss of the entire chromosome 22 in the tumor, but not in the blood sample. WGS confirmed previously unreported BRCA2 mutations, 6q loss, and 14q acquisition during tumor progression, but no other significant findings associated with tumor progression. The present case is discussed with reference to a systematic review of previous reports of AT/RT associated with PMS. PMS patients with ring chromosome 22 should be carefully followed up for AT/RT occurrence.
期刊介绍:
Brain Tumor Pathology is the official journal of the Japan Society of Brain Tumor Pathology. This international journal documents the latest research and topical debate in all clinical and experimental fields relating to brain tumors, especially brain tumor pathology. The journal has been published since 1983 and has been recognized worldwide as a unique journal of high quality. The journal welcomes the submission of manuscripts from any country. Membership in the society is not a prerequisite for submission. The journal publishes original articles, case reports, rapid short communications, instructional lectures, review articles, letters to the editor, and topics.Review articles and Topics may be recommended at the annual meeting of the Japan Society of Brain Tumor Pathology. All contributions should be aimed at promoting international scientific collaboration.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


