{"title":"Binding properties of the anti-TB drugs bedaquiline and TBAJ-876 to a mycobacterial F-ATP synthase","authors":"Alexander Krah , Gerhard Grüber , Peter J. Bond","doi":"10.1016/j.crstbi.2022.09.001","DOIUrl":null,"url":null,"abstract":"<div><p>Tuberculosis (TB), the deadly disease caused by <em>Mycobacterium tuberculosis</em> (<em>Mtb</em>), kills more people worldwide than any other bacterial infectious disease. There has been a recent resurgence of TB drug discovery activities, resulting in the identification of a number of novel enzyme inhibitors. Many of these inhibitors target the electron transport chain complexes and the F<sub>1</sub>F<sub>O</sub>-ATP synthase; these enzymes represent new target spaces for drug discovery, since the generation of ATP is essential for the bacterial pathogen's physiology, persistence, and pathogenicity. The anti-TB drug bedaquiline (BDQ) targets the <em>Mtb</em> F-ATP synthase and is used as salvage therapy against this disease. Medicinal chemistry efforts to improve the physio-chemical properties of BDQ resulted in the discovery of 3,5-dialkoxypyridine (DARQ) analogs to which TBAJ-876 belongs. TBAJ-876, a clinical development candidate, shows attractive <em>in vitro</em> and <em>in vivo</em> antitubercular activity. Both BDQ and TBAJ-876 inhibit the mycobacterial F<sub>1</sub>F<sub>O</sub>-ATP synthase by stopping rotation of the <em>c</em>-ring turbine within the F<sub>O</sub> domain, thereby preventing proton translocation and ATP synthesis to occur. While structural data for the BDQ bound state are available, no structural information about TBAJ-876 binding have been described. In this study, we show how TBAJ-876 binds to the F<sub>O</sub> domain of the <em>M. smegmatis</em> F<sub>1</sub>F<sub>O</sub>-ATP synthase. We further calculate the binding free energy of both drugs bound to their target and predict an increased affinity of TBAJ-876 for the F<sub>O</sub> domain. This approach will be useful in future efforts to design new and highly potent DARQ analogs targeting F-ATP synthases of <em>Mtb</em>, nontuberculosis mycobacteria (NTM) as well as the <em>M. leprosis</em> complex.</p></div>","PeriodicalId":10870,"journal":{"name":"Current Research in Structural Biology","volume":"4 ","pages":"Pages 278-284"},"PeriodicalIF":2.7000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9516385/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Research in Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2665928X22000253","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 4
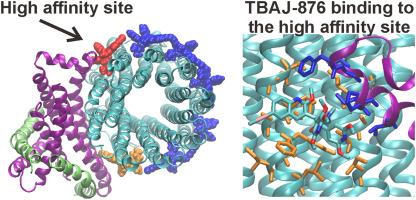
Abstract
Tuberculosis (TB), the deadly disease caused by Mycobacterium tuberculosis (Mtb), kills more people worldwide than any other bacterial infectious disease. There has been a recent resurgence of TB drug discovery activities, resulting in the identification of a number of novel enzyme inhibitors. Many of these inhibitors target the electron transport chain complexes and the F1FO-ATP synthase; these enzymes represent new target spaces for drug discovery, since the generation of ATP is essential for the bacterial pathogen's physiology, persistence, and pathogenicity. The anti-TB drug bedaquiline (BDQ) targets the Mtb F-ATP synthase and is used as salvage therapy against this disease. Medicinal chemistry efforts to improve the physio-chemical properties of BDQ resulted in the discovery of 3,5-dialkoxypyridine (DARQ) analogs to which TBAJ-876 belongs. TBAJ-876, a clinical development candidate, shows attractive in vitro and in vivo antitubercular activity. Both BDQ and TBAJ-876 inhibit the mycobacterial F1FO-ATP synthase by stopping rotation of the c-ring turbine within the FO domain, thereby preventing proton translocation and ATP synthesis to occur. While structural data for the BDQ bound state are available, no structural information about TBAJ-876 binding have been described. In this study, we show how TBAJ-876 binds to the FO domain of the M. smegmatis F1FO-ATP synthase. We further calculate the binding free energy of both drugs bound to their target and predict an increased affinity of TBAJ-876 for the FO domain. This approach will be useful in future efforts to design new and highly potent DARQ analogs targeting F-ATP synthases of Mtb, nontuberculosis mycobacteria (NTM) as well as the M. leprosis complex.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


