{"title":"Experiences of a Single Center in One Hundred Ninety-Four Adult Patients With Langerhans Cell Histiocytosis.","authors":"Claus Doberauer, Christoph Bornemann","doi":"10.14740/jh1020","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Langerhans cell histiocytosis (LCH) is a rare inflammatory myeloid neoplasia belonging to the group of histiocytoses. Inflammatory tissue destruction with fibrosis can result in dysfunction in any organ. Our evaluation aimed to collect information on characteristics, courses, and therapeutic options of this rare disease pattern in adult patients with conclusions on prognostic factors and follow-up management.</p><p><strong>Methods: </strong>The medical records of 194 adult patients with histologically confirmed LCH were evaluated in this retrospective study. Patients were treated at the Protestant Clinics in Gelsenkirchen from 2000 to 2014 and St. Franziskus-Hospital in Cologne until 2020.</p><p><strong>Results: </strong>The median age of onset was 38 years (18 to 79 years). In 65.5% of patients, only one organ was primarily involved, and in 34.5% of cases, multiple organs were involved. The skeleton, lungs, and skin were most commonly affected. In 15.5% of patients, pituitary insufficiency existed years before or at the time of diagnosis. The follow-up time of patients from the time of histologic diagnosis ranged from 6 to 408 months (median 49 months). Four patients died from sequelae of their underlying histiocytic disease. Irreversible late sequelae due to disease or therapy were detectable in 34% of patients. In 25.3% of the patients, the course of the disease could be controlled initially, but with the proviso of no smoking in case of lung involvement. Specific therapeutic measures such as surgery for solitary osteolysis, radiotherapy of osseous and cerebral manifestations, immunotherapy especially for lung and skin involvement, and chemotherapy for multisystem disease were primarily required in 74.7% of patients. As a result, 27.3% of all patients reached the nonactive stage. Of these, 26.4% had reactivation during the follow-up period. Of the remaining patients with continued active disease, 51.1% showed disease progression during follow-up.</p><p><strong>Conclusions: </strong>Standardized diagnostics are required to capture the clinical picture. Due to the variable course, it is often sufficient to initially control with obligatory smoking cessation in case of pulmonary involvement. Follow-up examinations should be predominantly symptom-oriented with attention to possible late sequelae.</p>","PeriodicalId":15964,"journal":{"name":"Journal of hematology","volume":"11 4","pages":"131-141"},"PeriodicalIF":1.3000,"publicationDate":"2022-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/72/12/jh-11-131.PMC9451550.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14740/jh1020","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/8/30 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
Background: Langerhans cell histiocytosis (LCH) is a rare inflammatory myeloid neoplasia belonging to the group of histiocytoses. Inflammatory tissue destruction with fibrosis can result in dysfunction in any organ. Our evaluation aimed to collect information on characteristics, courses, and therapeutic options of this rare disease pattern in adult patients with conclusions on prognostic factors and follow-up management.
Methods: The medical records of 194 adult patients with histologically confirmed LCH were evaluated in this retrospective study. Patients were treated at the Protestant Clinics in Gelsenkirchen from 2000 to 2014 and St. Franziskus-Hospital in Cologne until 2020.
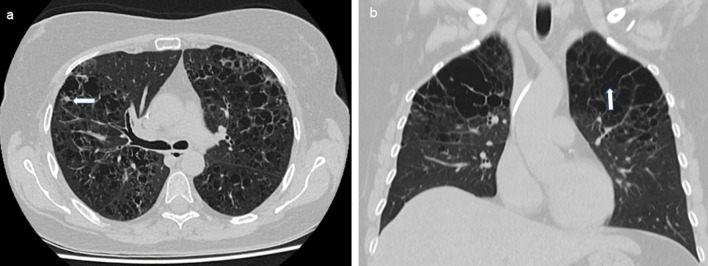
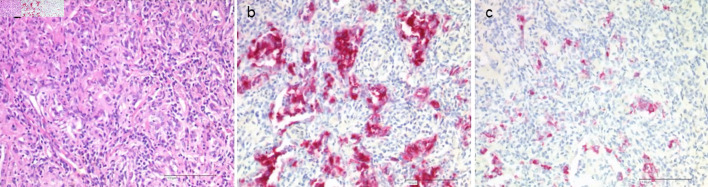
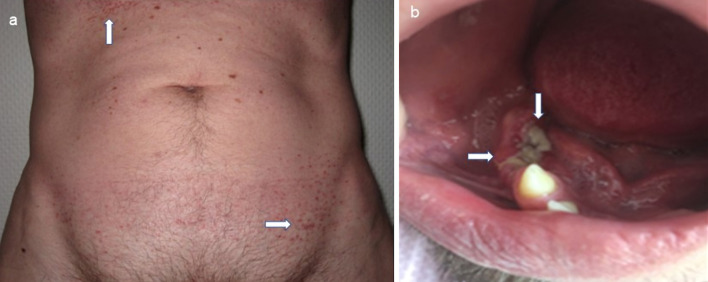
Results: The median age of onset was 38 years (18 to 79 years). In 65.5% of patients, only one organ was primarily involved, and in 34.5% of cases, multiple organs were involved. The skeleton, lungs, and skin were most commonly affected. In 15.5% of patients, pituitary insufficiency existed years before or at the time of diagnosis. The follow-up time of patients from the time of histologic diagnosis ranged from 6 to 408 months (median 49 months). Four patients died from sequelae of their underlying histiocytic disease. Irreversible late sequelae due to disease or therapy were detectable in 34% of patients. In 25.3% of the patients, the course of the disease could be controlled initially, but with the proviso of no smoking in case of lung involvement. Specific therapeutic measures such as surgery for solitary osteolysis, radiotherapy of osseous and cerebral manifestations, immunotherapy especially for lung and skin involvement, and chemotherapy for multisystem disease were primarily required in 74.7% of patients. As a result, 27.3% of all patients reached the nonactive stage. Of these, 26.4% had reactivation during the follow-up period. Of the remaining patients with continued active disease, 51.1% showed disease progression during follow-up.
Conclusions: Standardized diagnostics are required to capture the clinical picture. Due to the variable course, it is often sufficient to initially control with obligatory smoking cessation in case of pulmonary involvement. Follow-up examinations should be predominantly symptom-oriented with attention to possible late sequelae.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


