Jaydip Ray Chaudhuri, Jui Jade Bagul, Alluri Swathi, Bhim Sen Singhal, N Chakradhar Reddy, Kiran Kumar Vallam
{"title":"Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease Presenting as Intracranial Hypertension: A Case Report.","authors":"Jaydip Ray Chaudhuri, Jui Jade Bagul, Alluri Swathi, Bhim Sen Singhal, N Chakradhar Reddy, Kiran Kumar Vallam","doi":"10.1212/NXI.0000000000200020","DOIUrl":null,"url":null,"abstract":"<p><p>The production of autoantibodies against myelin oligodendrocyte glycoprotein (MOG) can cause a spectrum of autoimmune disorders, including optic neuritis, transverse myelitis, brainstem encephalitis, and acute disseminated encephalomyelitis. In this study, we present the case of a 19-year-old woman with an unusual clinical presentation of intracranial hypertension (IH) and bilateral papilledema. The patient presented with symptoms of increased intracranial pressure, which followed a relapsing, remitting course over several months. Serial CSF studies showed an increased opening pressure during clinical relapses. The CSF and serum tested positive for MOG immunoglobulin G antibodies. Contrast-enhanced MRI of the brain showed mild meningeal enhancement in the left parietal region with subtle underlying cortical hyperintensities, indicating possible fluid-attenuated inversion recovery variable unilateral enhancement of the leptomeninges. The patient responded well to immunosuppressive therapy using rituximab. The presentation of MOG antibody-associated disease (MOGAD) as IH without optic neuritis is rare. This report presents the first description of a relapsing remitting course presenting each time with only symptoms of raised intracranial pressure, without developing any typical clinical manifestations of MOGAD.</p>","PeriodicalId":520720,"journal":{"name":"Neurology(R) neuroimmunology & neuroinflammation","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2022-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7f/3f/NXI-2022-200026.PMC9581460.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology(R) neuroimmunology & neuroinflammation","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXI.0000000000200020","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/11/1 0:00:00","PubModel":"Print","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
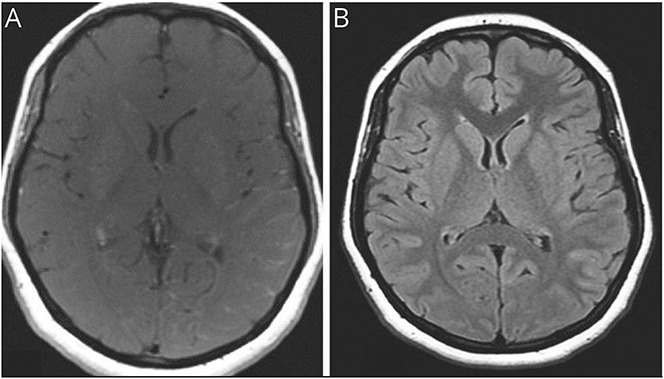
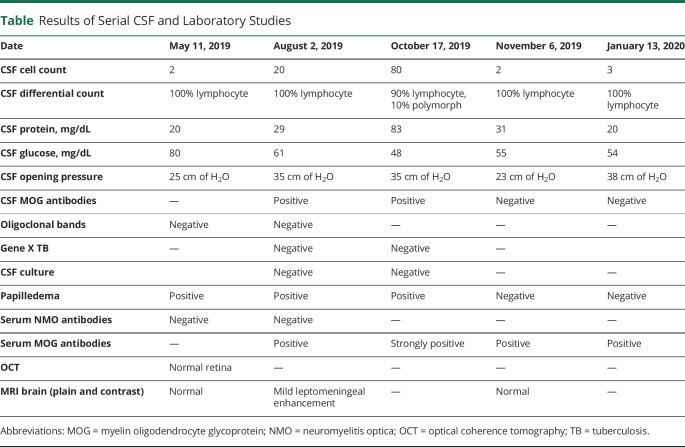
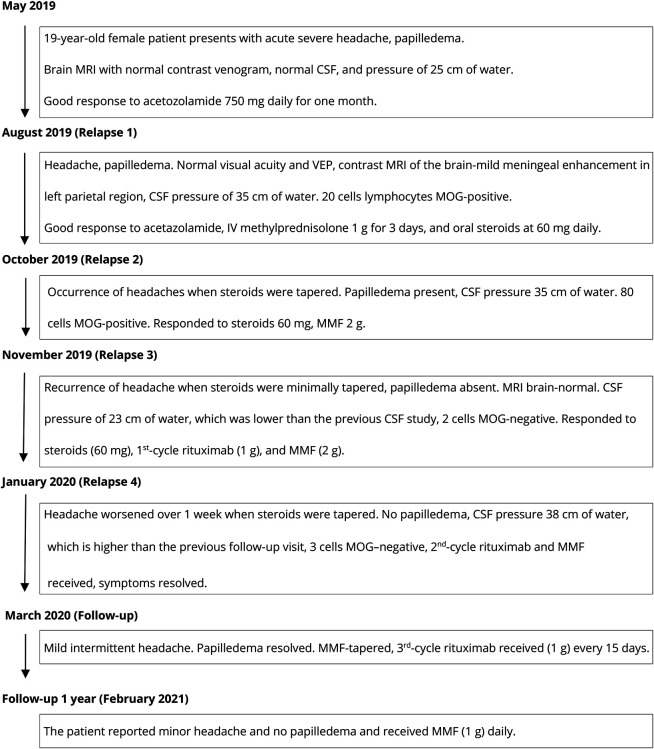
The production of autoantibodies against myelin oligodendrocyte glycoprotein (MOG) can cause a spectrum of autoimmune disorders, including optic neuritis, transverse myelitis, brainstem encephalitis, and acute disseminated encephalomyelitis. In this study, we present the case of a 19-year-old woman with an unusual clinical presentation of intracranial hypertension (IH) and bilateral papilledema. The patient presented with symptoms of increased intracranial pressure, which followed a relapsing, remitting course over several months. Serial CSF studies showed an increased opening pressure during clinical relapses. The CSF and serum tested positive for MOG immunoglobulin G antibodies. Contrast-enhanced MRI of the brain showed mild meningeal enhancement in the left parietal region with subtle underlying cortical hyperintensities, indicating possible fluid-attenuated inversion recovery variable unilateral enhancement of the leptomeninges. The patient responded well to immunosuppressive therapy using rituximab. The presentation of MOG antibody-associated disease (MOGAD) as IH without optic neuritis is rare. This report presents the first description of a relapsing remitting course presenting each time with only symptoms of raised intracranial pressure, without developing any typical clinical manifestations of MOGAD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


