Martina Rueca, Emanuela Giombini, Francesco Messina, Barbara Bartolini, Antonino Di Caro, Maria Rosaria Capobianchi, Cesare Em Gruber
{"title":"The Easy-to-Use SARS-CoV-2 Assembler for Genome Sequencing: Development Study.","authors":"Martina Rueca, Emanuela Giombini, Francesco Messina, Barbara Bartolini, Antonino Di Caro, Maria Rosaria Capobianchi, Cesare Em Gruber","doi":"10.2196/31536","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Early sequencing and quick analysis of the SARS-CoV-2 genome have contributed to the understanding of the dynamics of COVID-19 epidemics and in designing countermeasures at a global level.</p><p><strong>Objective: </strong>Amplicon-based next-generation sequencing (NGS) methods are widely used to sequence the SARS-CoV-2 genome and to identify novel variants that are emerging in rapid succession as well as harboring multiple deletions and amino acid-changing mutations.</p><p><strong>Methods: </strong>To facilitate the analysis of NGS sequencing data obtained from amplicon-based sequencing methods, here, we propose an easy-to-use SARS-CoV-2 genome assembler: the Easy-to-use SARS-CoV-2 Assembler (ESCA) pipeline.</p><p><strong>Results: </strong>Our results have shown that ESCA could perform high-quality genome assembly from Ion Torrent and Illumina raw data and help the user in easily correct low-coverage regions. Moreover, ESCA includes the possibility of comparing assembled genomes of multisample runs through an easy table format.</p><p><strong>Conclusions: </strong>In conclusion, ESCA automatically furnished a variant table output file, fundamental to rapidly recognizing variants of interest. Our pipeline could be a useful method for obtaining a complete, rapid, and accurate analysis even with minimal knowledge in bioinformatics.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":" ","pages":"e31536"},"PeriodicalIF":0.0000,"publicationDate":"2022-03-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8924907/pdf/","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/31536","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
Abstract
Background: Early sequencing and quick analysis of the SARS-CoV-2 genome have contributed to the understanding of the dynamics of COVID-19 epidemics and in designing countermeasures at a global level.
Objective: Amplicon-based next-generation sequencing (NGS) methods are widely used to sequence the SARS-CoV-2 genome and to identify novel variants that are emerging in rapid succession as well as harboring multiple deletions and amino acid-changing mutations.
Methods: To facilitate the analysis of NGS sequencing data obtained from amplicon-based sequencing methods, here, we propose an easy-to-use SARS-CoV-2 genome assembler: the Easy-to-use SARS-CoV-2 Assembler (ESCA) pipeline.
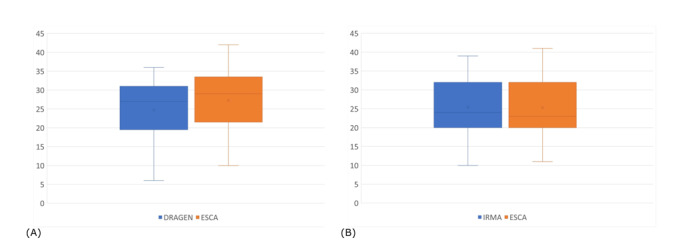
Results: Our results have shown that ESCA could perform high-quality genome assembly from Ion Torrent and Illumina raw data and help the user in easily correct low-coverage regions. Moreover, ESCA includes the possibility of comparing assembled genomes of multisample runs through an easy table format.
Conclusions: In conclusion, ESCA automatically furnished a variant table output file, fundamental to rapidly recognizing variants of interest. Our pipeline could be a useful method for obtaining a complete, rapid, and accurate analysis even with minimal knowledge in bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


