Antonio Muñoz-Mérida, Juan José González-Plaza, Andrés Cañada, Ana María Blanco, Maria del Carmen García-López, José Manuel Rodríguez, Laia Pedrola, M Dolores Sicardo, M Luisa Hernández, Raúl De la Rosa, Angjelina Belaj, Mayte Gil-Borja, Francisco Luque, José Manuel Martínez-Rivas, David G Pisano, Oswaldo Trelles, Victoriano Valpuesta, Carmen R Beuzón
{"title":"De novo assembly and functional annotation of the olive (Olea europaea) transcriptome.","authors":"Antonio Muñoz-Mérida, Juan José González-Plaza, Andrés Cañada, Ana María Blanco, Maria del Carmen García-López, José Manuel Rodríguez, Laia Pedrola, M Dolores Sicardo, M Luisa Hernández, Raúl De la Rosa, Angjelina Belaj, Mayte Gil-Borja, Francisco Luque, José Manuel Martínez-Rivas, David G Pisano, Oswaldo Trelles, Victoriano Valpuesta, Carmen R Beuzón","doi":"10.1093/dnares/dss036","DOIUrl":null,"url":null,"abstract":"<p><p>Olive breeding programmes are focused on selecting for traits as short juvenile period, plant architecture suited for mechanical harvest, or oil characteristics, including fatty acid composition, phenolic, and volatile compounds to suit new markets. Understanding the molecular basis of these characteristics and improving the efficiency of such breeding programmes require the development of genomic information and tools. However, despite its economic relevance, genomic information on olive or closely related species is still scarce. We have applied Sanger and 454 pyrosequencing technologies to generate close to 2 million reads from 12 cDNA libraries obtained from the Picual, Arbequina, and Lechin de Sevilla cultivars and seedlings from a segregating progeny of a Picual × Arbequina cross. The libraries include fruit mesocarp and seeds at three relevant developmental stages, young stems and leaves, active juvenile and adult buds as well as dormant buds, and juvenile and adult roots. The reads were assembled by library or tissue and then assembled together into 81 020 unigenes with an average size of 496 bases. Here, we report their assembly and their functional annotation.</p>","PeriodicalId":11212,"journal":{"name":"DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes","volume":" ","pages":"93-108"},"PeriodicalIF":0.0000,"publicationDate":"2013-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1093/dnares/dss036","citationCount":"82","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/dnares/dss036","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/7 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 82
Abstract
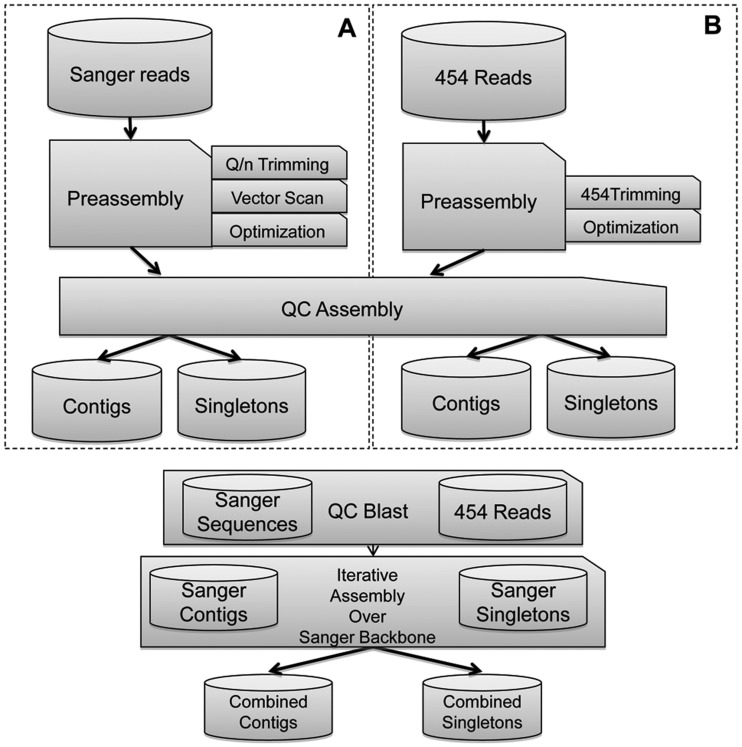
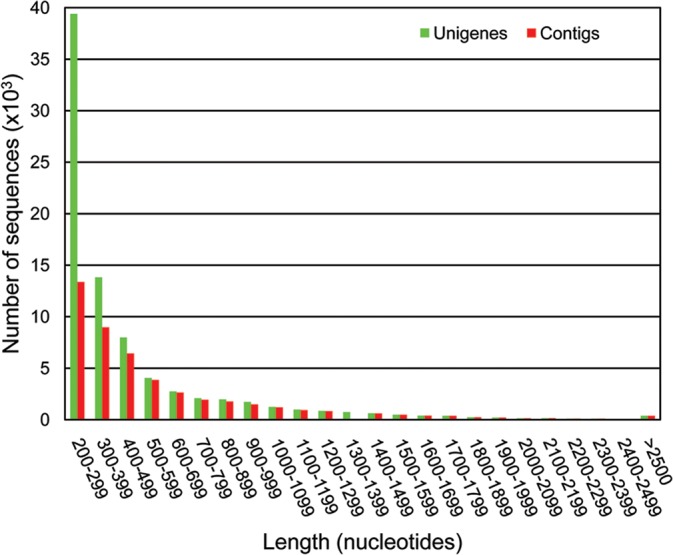
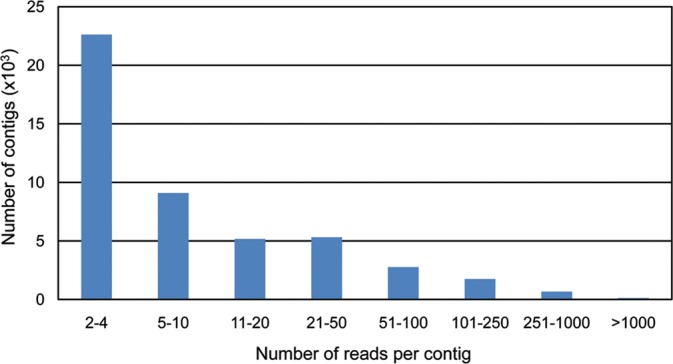
Olive breeding programmes are focused on selecting for traits as short juvenile period, plant architecture suited for mechanical harvest, or oil characteristics, including fatty acid composition, phenolic, and volatile compounds to suit new markets. Understanding the molecular basis of these characteristics and improving the efficiency of such breeding programmes require the development of genomic information and tools. However, despite its economic relevance, genomic information on olive or closely related species is still scarce. We have applied Sanger and 454 pyrosequencing technologies to generate close to 2 million reads from 12 cDNA libraries obtained from the Picual, Arbequina, and Lechin de Sevilla cultivars and seedlings from a segregating progeny of a Picual × Arbequina cross. The libraries include fruit mesocarp and seeds at three relevant developmental stages, young stems and leaves, active juvenile and adult buds as well as dormant buds, and juvenile and adult roots. The reads were assembled by library or tissue and then assembled together into 81 020 unigenes with an average size of 496 bases. Here, we report their assembly and their functional annotation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


