Clelia Peano, Alessandro Pietrelli, Clarissa Consolandi, Elio Rossi, Luca Petiti, Letizia Tagliabue, Gianluca De Bellis, Paolo Landini
{"title":"An efficient rRNA removal method for RNA sequencing in GC-rich bacteria.","authors":"Clelia Peano, Alessandro Pietrelli, Clarissa Consolandi, Elio Rossi, Luca Petiti, Letizia Tagliabue, Gianluca De Bellis, Paolo Landini","doi":"10.1186/2042-5783-3-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Next generation sequencing (NGS) technologies have revolutionized gene expression studies and functional genomics analysis. However, further improvement of RNA sequencing protocols is still desirable, in order to reduce NGS costs and to increase its accuracy. In bacteria, a major problem in RNA sequencing is the abundance of ribosomal RNA (rRNA), which accounts for 95-98% of total RNA and can therefore hinder sufficient coverage of mRNA, the main focus of transcriptomic studies. Thus, efficient removal of rRNA is necessary to achieve optimal coverage, good detection sensitivity and reliable results. An additional challenge is presented by microorganisms with GC-rich genomes, in which rRNA removal is less efficient.</p><p><strong>Results: </strong>In this work, we tested two commercial kits for rRNA removal, either alone or in combination, on Burkholderia thailandensis. This bacterium, chosen as representative of the important Burkholderia genus, which includes both pathogenic and environmental bacteria, has a rather large (6.72 Mb) and GC-rich (67.7%) genome. Each enriched mRNA sample was sequenced through paired-end Illumina GAIIx run in duplicate, yielding between 10 and 40 million reads. We show that combined treatment with both kits allows an mRNA enrichment of more than 238-fold, enabling the sequencing of almost all (more than 90%) B. thailandensis transcripts from less than 10 million reads, without introducing any bias in mRNA relative abundance, thus preserving differential expression profile.</p><p><strong>Conclusions: </strong>The mRNA enrichment protocol presented in this work leads to an increase in detection sensitivity up to 770% compared to total RNA; such increased sensitivity allows for a corresponding reduction in the number of sequencing reads necessary for the complete analysis of whole transcriptome expression profiling. Thus we can conclude that the MICROBExpress/Ovation combined rRNA removal method could be suitable for RNA sequencing of whole transcriptomes of microorganisms with high GC content and complex genomes enabling at the same time an important scaling down of sequencing costs.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":" ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-3-1","citationCount":"55","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-3-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 55
Abstract
Unlabelled:
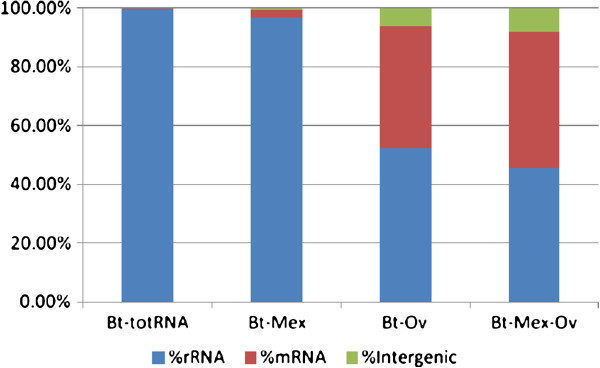
Background: Next generation sequencing (NGS) technologies have revolutionized gene expression studies and functional genomics analysis. However, further improvement of RNA sequencing protocols is still desirable, in order to reduce NGS costs and to increase its accuracy. In bacteria, a major problem in RNA sequencing is the abundance of ribosomal RNA (rRNA), which accounts for 95-98% of total RNA and can therefore hinder sufficient coverage of mRNA, the main focus of transcriptomic studies. Thus, efficient removal of rRNA is necessary to achieve optimal coverage, good detection sensitivity and reliable results. An additional challenge is presented by microorganisms with GC-rich genomes, in which rRNA removal is less efficient.
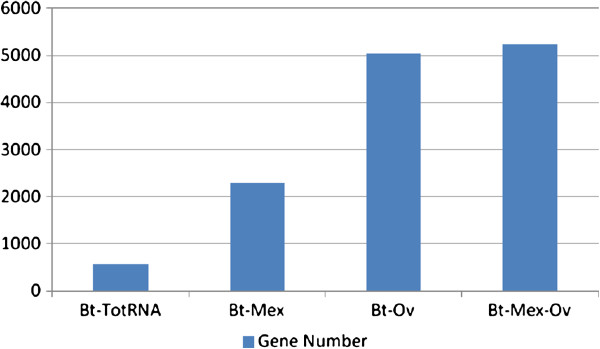
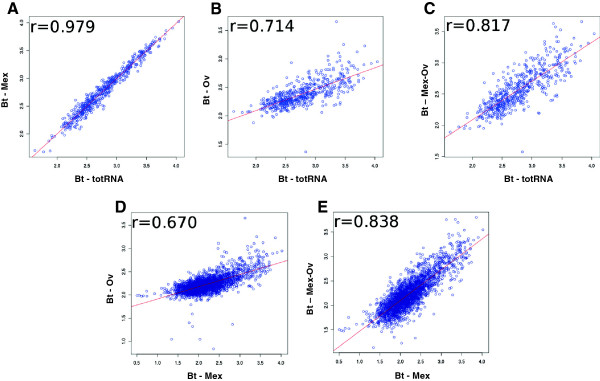
Results: In this work, we tested two commercial kits for rRNA removal, either alone or in combination, on Burkholderia thailandensis. This bacterium, chosen as representative of the important Burkholderia genus, which includes both pathogenic and environmental bacteria, has a rather large (6.72 Mb) and GC-rich (67.7%) genome. Each enriched mRNA sample was sequenced through paired-end Illumina GAIIx run in duplicate, yielding between 10 and 40 million reads. We show that combined treatment with both kits allows an mRNA enrichment of more than 238-fold, enabling the sequencing of almost all (more than 90%) B. thailandensis transcripts from less than 10 million reads, without introducing any bias in mRNA relative abundance, thus preserving differential expression profile.
Conclusions: The mRNA enrichment protocol presented in this work leads to an increase in detection sensitivity up to 770% compared to total RNA; such increased sensitivity allows for a corresponding reduction in the number of sequencing reads necessary for the complete analysis of whole transcriptome expression profiling. Thus we can conclude that the MICROBExpress/Ovation combined rRNA removal method could be suitable for RNA sequencing of whole transcriptomes of microorganisms with high GC content and complex genomes enabling at the same time an important scaling down of sequencing costs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


