Subit Barua, Sara Berger, Elaine M Pereira, Vaidehi Jobanputra
{"title":"Expanding the phenotype of <i>ATP6AP1</i> deficiency.","authors":"Subit Barua, Sara Berger, Elaine M Pereira, Vaidehi Jobanputra","doi":"10.1101/mcs.a006195","DOIUrl":null,"url":null,"abstract":"<p><p>Vacuolar ATPases (V-ATPases) are large multisubunit proton pumps conserved among all eukaryotic cells that are involved in diverse functions including acidification of membrane-bound intracellular compartments. The <i>ATP6AP1</i> gene encodes an accessory subunit of the vacuolar (V)-ATPase protein pump. Pathogenic variants in <i>ATP6AP1</i> have been described in association with a congenital disorder of glycosylation (CDG), which are highly variable, but often characterized by immunodeficiency, hepatopathy, and neurologic manifestations. Although the most striking and common clinical feature is hepatopathy, the phenotypic and genotypic spectrum of <i>ATP6AP1-CDG</i> continues to expand. Here, we report identical twins who presented with acute liver failure and jaundice. Prenatal features included cystic hygroma, atrial septal defect, and ventriculomegaly. Postnatal features included pectus carinatum, connective tissue abnormalities, and hypospadias. Whole-exome sequencing (WES) revealed a novel de novo in-frame deletion in the <i>ATP6AP1</i> gene (c.230_232delACT;p.Tyr77del). Although both twins have the commonly reported clinical feature of hepatopathy seen in other individuals with <i>ATP6AP1-CDG</i>-related disorder, they do not have neurological sequelae. This report expands the phenotypic spectrum of <i>ATP6AP1-CDG-</i>related disorder with both probands exhibiting unique prenatal and postnatal features, including fetal ventriculomegaly, umbilical hernia, pectus carinatum, micropenis, and hypospadias. Furthermore, this case affirms that neurological features described in the initial case series on <i>ATP6AP1-CDG</i> do not appear to be central, whereas the prenatal and connective tissue manifestations may be more common than previously thought. This emphasizes the importance of long-term clinical follow-up and variant interpretation using current updated recommendations.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-06-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c0/b9/MCS006195Bar.PMC9235842.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006195","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 3
Abstract
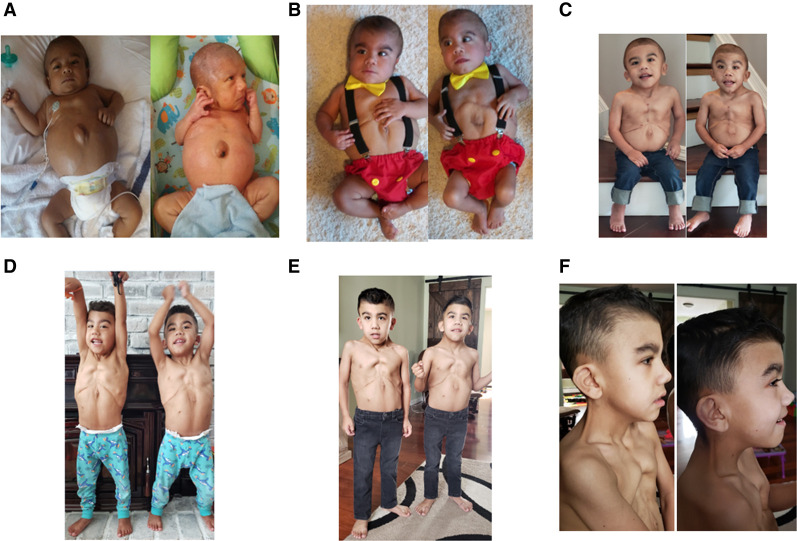
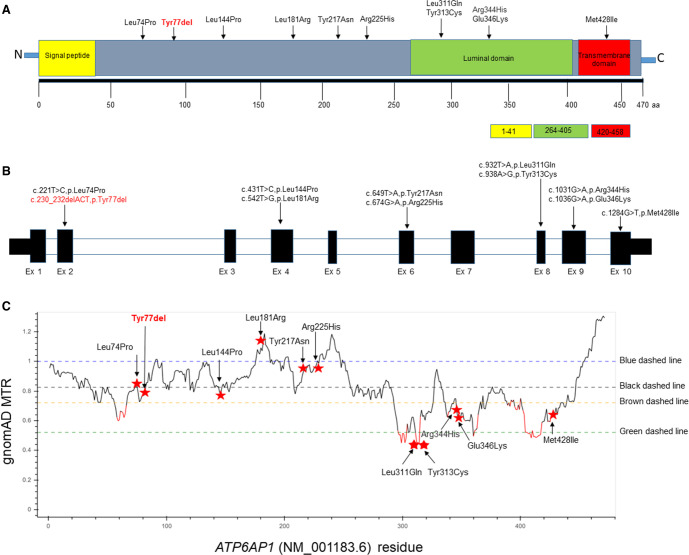
Vacuolar ATPases (V-ATPases) are large multisubunit proton pumps conserved among all eukaryotic cells that are involved in diverse functions including acidification of membrane-bound intracellular compartments. The ATP6AP1 gene encodes an accessory subunit of the vacuolar (V)-ATPase protein pump. Pathogenic variants in ATP6AP1 have been described in association with a congenital disorder of glycosylation (CDG), which are highly variable, but often characterized by immunodeficiency, hepatopathy, and neurologic manifestations. Although the most striking and common clinical feature is hepatopathy, the phenotypic and genotypic spectrum of ATP6AP1-CDG continues to expand. Here, we report identical twins who presented with acute liver failure and jaundice. Prenatal features included cystic hygroma, atrial septal defect, and ventriculomegaly. Postnatal features included pectus carinatum, connective tissue abnormalities, and hypospadias. Whole-exome sequencing (WES) revealed a novel de novo in-frame deletion in the ATP6AP1 gene (c.230_232delACT;p.Tyr77del). Although both twins have the commonly reported clinical feature of hepatopathy seen in other individuals with ATP6AP1-CDG-related disorder, they do not have neurological sequelae. This report expands the phenotypic spectrum of ATP6AP1-CDG-related disorder with both probands exhibiting unique prenatal and postnatal features, including fetal ventriculomegaly, umbilical hernia, pectus carinatum, micropenis, and hypospadias. Furthermore, this case affirms that neurological features described in the initial case series on ATP6AP1-CDG do not appear to be central, whereas the prenatal and connective tissue manifestations may be more common than previously thought. This emphasizes the importance of long-term clinical follow-up and variant interpretation using current updated recommendations.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


