Willem J de Lange, Adrian C Grimes, Laura F Hegge, J Carter Ralphe
{"title":"Ablation of cardiac myosin-binding protein-C accelerates contractile kinetics in engineered cardiac tissue.","authors":"Willem J de Lange, Adrian C Grimes, Laura F Hegge, J Carter Ralphe","doi":"10.1085/jgp.201210837","DOIUrl":null,"url":null,"abstract":"<p><p>Hypertrophic cardiomyopathy (HCM) caused by mutations in cardiac myosin-binding protein-C (cMyBP-C) is a heterogenous disease in which the phenotypic presentation is influenced by genetic, environmental, and developmental factors. Though mouse models have been used extensively to study the contractile effects of cMyBP-C ablation, early postnatal hypertrophic and dilatory remodeling may overshadow primary contractile defects. The use of a murine engineered cardiac tissue (mECT) model of cMyBP-C ablation in the present study permits delineation of the primary contractile kinetic abnormalities in an intact tissue model under mechanical loading conditions in the absence of confounding remodeling events. We generated mechanically integrated mECT using isolated postnatal day 1 mouse cardiac cells from both wild-type (WT) and cMyBP-C-null hearts. After culturing for 1 wk to establish coordinated spontaneous contraction, we measured twitch force and Ca(2+) transients at 37°C during pacing at 6 and 9 Hz, with and without dobutamine. Compared with WT, the cMyBP-C-null mECT demonstrated faster late contraction kinetics and significantly faster early relaxation kinetics with no difference in Ca(2+) transient kinetics. Strikingly, the ability of cMyBP-C-null mECT to increase contractile kinetics in response to adrenergic stimulation and increased pacing frequency were severely impaired. We conclude that cMyBP-C ablation results in constitutively accelerated contractile kinetics with preserved peak force with minimal contractile kinetic reserve. These functional abnormalities precede the development of the hypertrophic phenotype and do not result from alterations in Ca(2+) transient kinetics, suggesting that alterations in contractile velocity may serve as the primary functional trigger for the development of hypertrophy in this model of HCM. Our findings strongly support a mechanism in which cMyBP-C functions as a physiological brake on contraction by positioning myosin heads away from the thin filament, a constraint which is removed upon adrenergic stimulation or cMyBP-C ablation.</p>","PeriodicalId":173753,"journal":{"name":"The Journal of General Physiology","volume":" ","pages":"73-84"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1085/jgp.201210837","citationCount":"30","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of General Physiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1085/jgp.201210837","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 30
Abstract
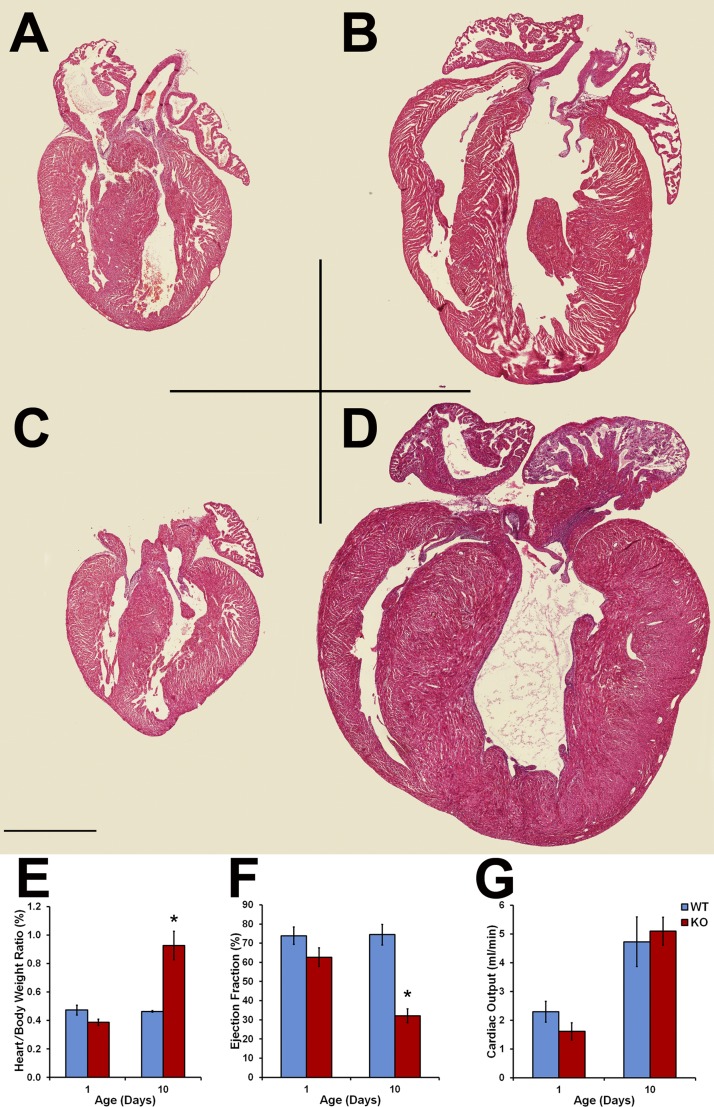
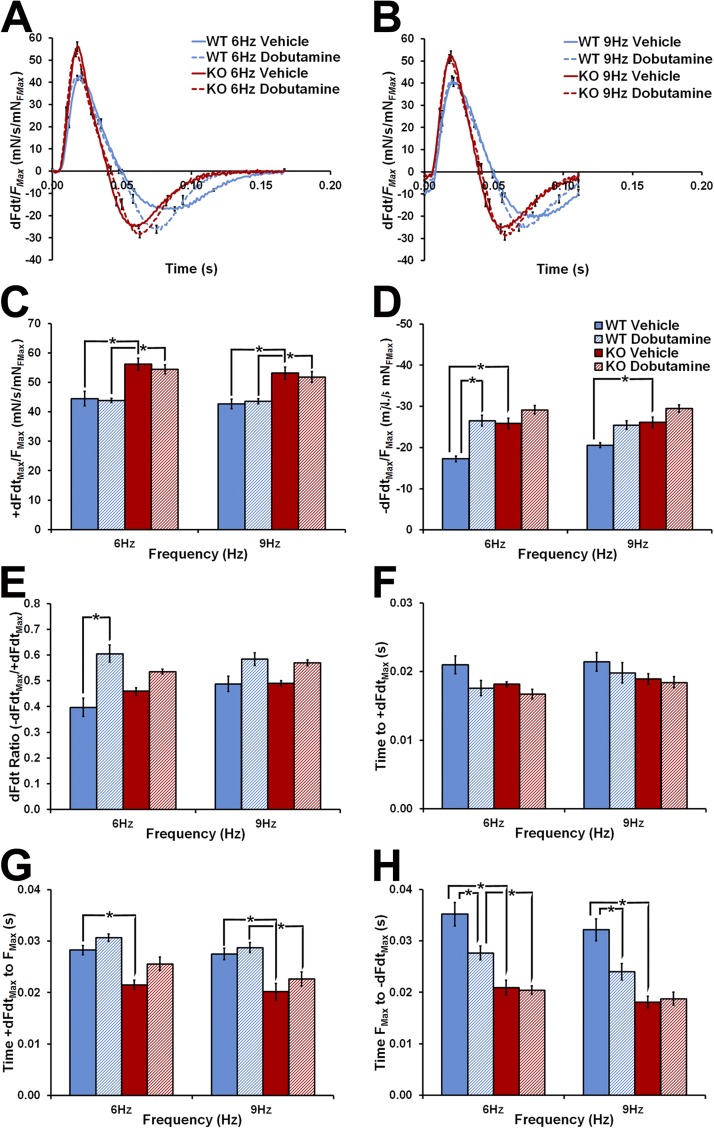
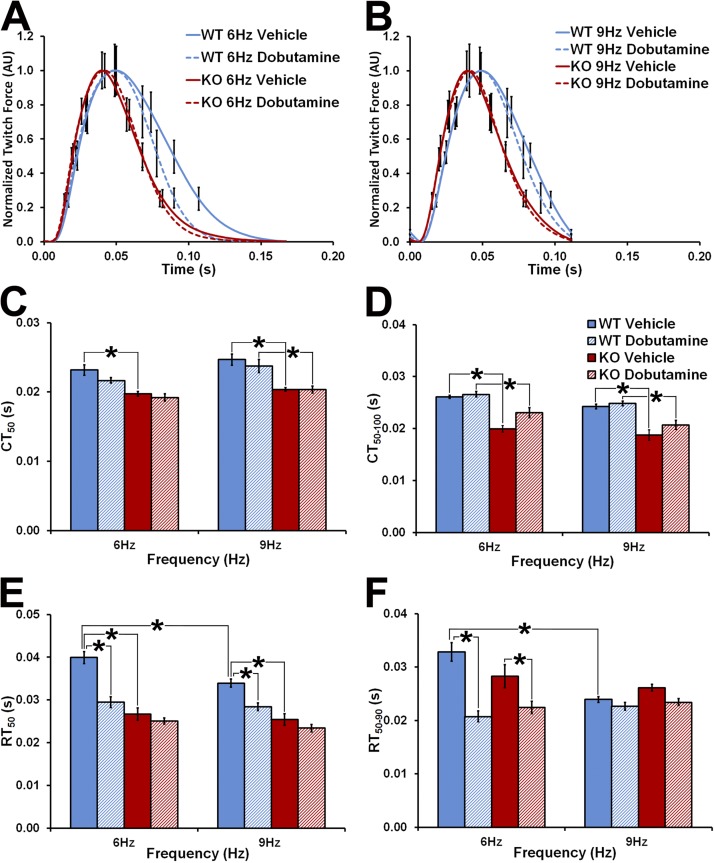
Hypertrophic cardiomyopathy (HCM) caused by mutations in cardiac myosin-binding protein-C (cMyBP-C) is a heterogenous disease in which the phenotypic presentation is influenced by genetic, environmental, and developmental factors. Though mouse models have been used extensively to study the contractile effects of cMyBP-C ablation, early postnatal hypertrophic and dilatory remodeling may overshadow primary contractile defects. The use of a murine engineered cardiac tissue (mECT) model of cMyBP-C ablation in the present study permits delineation of the primary contractile kinetic abnormalities in an intact tissue model under mechanical loading conditions in the absence of confounding remodeling events. We generated mechanically integrated mECT using isolated postnatal day 1 mouse cardiac cells from both wild-type (WT) and cMyBP-C-null hearts. After culturing for 1 wk to establish coordinated spontaneous contraction, we measured twitch force and Ca(2+) transients at 37°C during pacing at 6 and 9 Hz, with and without dobutamine. Compared with WT, the cMyBP-C-null mECT demonstrated faster late contraction kinetics and significantly faster early relaxation kinetics with no difference in Ca(2+) transient kinetics. Strikingly, the ability of cMyBP-C-null mECT to increase contractile kinetics in response to adrenergic stimulation and increased pacing frequency were severely impaired. We conclude that cMyBP-C ablation results in constitutively accelerated contractile kinetics with preserved peak force with minimal contractile kinetic reserve. These functional abnormalities precede the development of the hypertrophic phenotype and do not result from alterations in Ca(2+) transient kinetics, suggesting that alterations in contractile velocity may serve as the primary functional trigger for the development of hypertrophy in this model of HCM. Our findings strongly support a mechanism in which cMyBP-C functions as a physiological brake on contraction by positioning myosin heads away from the thin filament, a constraint which is removed upon adrenergic stimulation or cMyBP-C ablation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


