Uğur Güller, Şükrü Beydemir, Ömer İrfan Küfrevioğlu
{"title":"In vitro and in silico interactions of antiulcer, glucocorticoids and urological drugs on human carbonic anhydrase I and II isozymes","authors":"Uğur Güller, Şükrü Beydemir, Ömer İrfan Küfrevioğlu","doi":"10.1002/bdd.2309","DOIUrl":null,"url":null,"abstract":"<p>Carbonic anhydrases (CAs, Enzyme Commission 4.2.1.1) convert carbon dioxide to bicarbonate in metabolism and use Zn<sup>2+</sup> ions as a cofactor for their catalytic activity. The activators or inhibitors of CA-I and CA-II, which are the most abundant CA isozymes in erythrocytes, have pharmacological applications in medicine. So, investigation of drug-protein interaction of these isozymes is significant. On this basis, the objective of this study was to clarify the primer effects of widely used drugs on the activity of human CA-I and CA-II enzymes and elucidate the inhibition mechanism through molecular docking studies. For this aim isozymes were purified from human erythrocytes by affinity chromatography technique. Then inhibition profiles of antiulcer, glucocorticoids, and urological drugs were investigated. As a result, while budesonide had the highest inhibitory potency on hydratase activity of hCA-I with the IC<sub>50</sub> of 0.08 mM, levofloxacin showed the highest inhibition effect on hCA-II with the IC<sub>50</sub> of 0.886 mM. The most effective inhibitor on the esterase activity of isozymes was found as fluticasone propionate with the K<sub>i</sub> values of 0.0365 ± 0.016 mM and 0.054 ± 0.018 mM respectively. However, by molecular docking study, it was estimated that budesonide showed maximum inhibition potency for both isozymes with the free binding energy of −7.58 and −6.97 kcal/mol, respectively. Consequently, it was observed that some of the drugs studied did not show any inhibitory effect. Drug-enzyme interactions were also estimated by molecular docking. This study could contribute to the discovery of new drug candidates and as well as target proteins.</p>","PeriodicalId":8865,"journal":{"name":"Biopharmaceutics & Drug Disposition","volume":"43 2","pages":"47-56"},"PeriodicalIF":1.7000,"publicationDate":"2022-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biopharmaceutics & Drug Disposition","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bdd.2309","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 3
Abstract
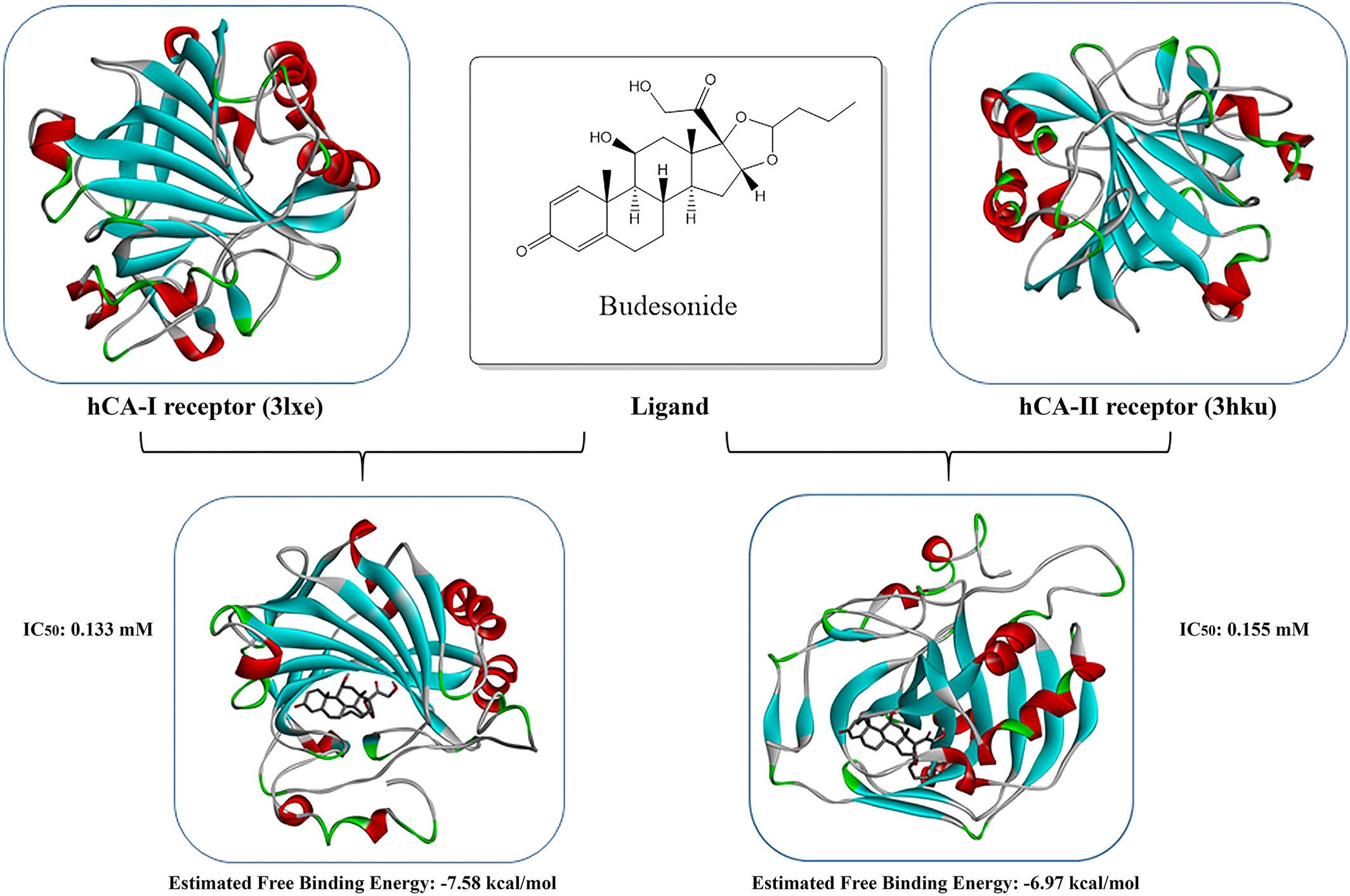
Carbonic anhydrases (CAs, Enzyme Commission 4.2.1.1) convert carbon dioxide to bicarbonate in metabolism and use Zn2+ ions as a cofactor for their catalytic activity. The activators or inhibitors of CA-I and CA-II, which are the most abundant CA isozymes in erythrocytes, have pharmacological applications in medicine. So, investigation of drug-protein interaction of these isozymes is significant. On this basis, the objective of this study was to clarify the primer effects of widely used drugs on the activity of human CA-I and CA-II enzymes and elucidate the inhibition mechanism through molecular docking studies. For this aim isozymes were purified from human erythrocytes by affinity chromatography technique. Then inhibition profiles of antiulcer, glucocorticoids, and urological drugs were investigated. As a result, while budesonide had the highest inhibitory potency on hydratase activity of hCA-I with the IC50 of 0.08 mM, levofloxacin showed the highest inhibition effect on hCA-II with the IC50 of 0.886 mM. The most effective inhibitor on the esterase activity of isozymes was found as fluticasone propionate with the Ki values of 0.0365 ± 0.016 mM and 0.054 ± 0.018 mM respectively. However, by molecular docking study, it was estimated that budesonide showed maximum inhibition potency for both isozymes with the free binding energy of −7.58 and −6.97 kcal/mol, respectively. Consequently, it was observed that some of the drugs studied did not show any inhibitory effect. Drug-enzyme interactions were also estimated by molecular docking. This study could contribute to the discovery of new drug candidates and as well as target proteins.
期刊介绍:
Biopharmaceutics & Drug Dispositionpublishes original review articles, short communications, and reports in biopharmaceutics, drug disposition, pharmacokinetics and pharmacodynamics, especially those that have a direct relation to the drug discovery/development and the therapeutic use of drugs. These includes:
- animal and human pharmacological studies that focus on therapeutic response. pharmacodynamics, and toxicity related to plasma and tissue concentrations of drugs and their metabolites,
- in vitro and in vivo drug absorption, distribution, metabolism, transport, and excretion studies that facilitate investigations related to the use of drugs in man
- studies on membrane transport and enzymes, including their regulation and the impact of pharmacogenomics on drug absorption and disposition,
- simulation and modeling in drug discovery and development
- theoretical treatises
- includes themed issues and reviews
and exclude manuscripts on
- bioavailability studies reporting only on simple PK parameters such as Cmax, tmax and t1/2 without mechanistic interpretation
- analytical methods

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


