{"title":"Translational research approaches to study pediatric polycystic kidney disease.","authors":"Max Christoph Liebau, Djalila Mekahli","doi":"10.1186/s40348-021-00131-x","DOIUrl":null,"url":null,"abstract":"<p><p>Polycystic kidney diseases (PKD) are severe forms of genetic kidney disorders. The two main types of PKD are autosomal recessive and autosomal dominant PKD (ARPKD, ADPKD). While ARPKD typically is a disorder of early childhood, patients with ADPKD often remain pauci-symptomatic until adulthood even though formation of cysts in the kidney already begins in children. There is clinical and genetic overlap between both entities with very variable clinical courses. Subgroups of very early onset ADPKD may for example clinically resemble ARPKD. The basis of the clinical variability in both forms of PKD is not well understood and there are also limited prediction markers for disease progression for daily clinical life or surrogate endpoints for clinical trials in ARPKD or early ADPKD.As targeted therapeutic approaches to slow disease progression in PKD are emerging, it is becoming more important to reliably identify patients at risk for rapid progression as they might benefit from early therapy. Over the past years regional, national and international data collections to jointly analyze the clinical courses of PKD patients have been set up. The clinical observations are complemented by genetic studies and biorepositories as well as basic science approaches to elucidate the underlying molecular mechanisms in the PKD field. These approaches may serve as a basis for the development of novel therapeutic interventions in specific subgroups of patients. In this article we summarize some of the recent developments in the field with a focus on kidney involvement in PKD during childhood and adolescence and findings obtained in pediatric cohorts.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"8 1","pages":"20"},"PeriodicalIF":2.4000,"publicationDate":"2021-12-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8660924/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-021-00131-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 1
Abstract
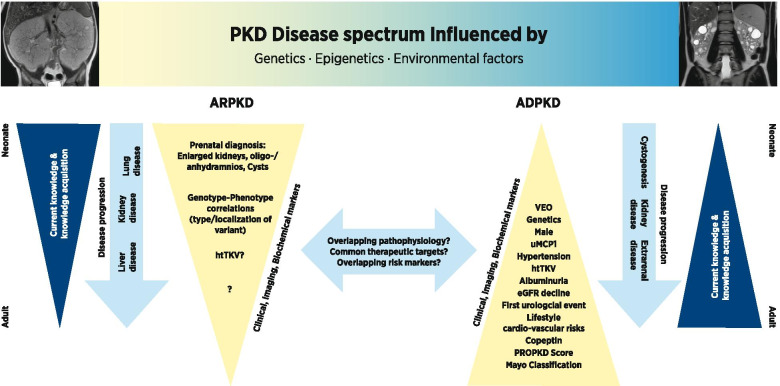
Polycystic kidney diseases (PKD) are severe forms of genetic kidney disorders. The two main types of PKD are autosomal recessive and autosomal dominant PKD (ARPKD, ADPKD). While ARPKD typically is a disorder of early childhood, patients with ADPKD often remain pauci-symptomatic until adulthood even though formation of cysts in the kidney already begins in children. There is clinical and genetic overlap between both entities with very variable clinical courses. Subgroups of very early onset ADPKD may for example clinically resemble ARPKD. The basis of the clinical variability in both forms of PKD is not well understood and there are also limited prediction markers for disease progression for daily clinical life or surrogate endpoints for clinical trials in ARPKD or early ADPKD.As targeted therapeutic approaches to slow disease progression in PKD are emerging, it is becoming more important to reliably identify patients at risk for rapid progression as they might benefit from early therapy. Over the past years regional, national and international data collections to jointly analyze the clinical courses of PKD patients have been set up. The clinical observations are complemented by genetic studies and biorepositories as well as basic science approaches to elucidate the underlying molecular mechanisms in the PKD field. These approaches may serve as a basis for the development of novel therapeutic interventions in specific subgroups of patients. In this article we summarize some of the recent developments in the field with a focus on kidney involvement in PKD during childhood and adolescence and findings obtained in pediatric cohorts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


