Simulations of x-ray absorption spectra for CO desorbing from Ru(0001) with transition-potential and time-dependent density functional theory approaches.
Gabriel L S Rodrigues, Elias Diesen, Johannes Voss, Patrick Norman, Lars G M Pettersson
{"title":"Simulations of x-ray absorption spectra for CO desorbing from Ru(0001) with transition-potential and time-dependent density functional theory approaches.","authors":"Gabriel L S Rodrigues, Elias Diesen, Johannes Voss, Patrick Norman, Lars G M Pettersson","doi":"10.1063/4.0000135","DOIUrl":null,"url":null,"abstract":"<p><p>The desorption of a carbon monoxide molecule from a Ru(0001) surface was studied by means of X-ray Absorption Spectra (XAS) computed with Transition Potential (TP-DFT) and Time Dependent (TD-DFT) DFT methods. By unraveling the evolution of the CO electronic structure upon desorption, we observed that at 2.3 Å from the surface, the CO molecule has already predominantly gas-phase character. While C 1s XAS is quite insensitive to changes in the C-O bond length, the O 1s excitation is very sensitive with the π* coming down in energy upon CO bond stretching, which competes with the increase in orbital energy due to the repulsive interaction with the metallic surface. We show in a systematic way that the TP-DFT method can describe the XAS rather well at the endpoints (chemisorbed and gas phase) but is affected by artificial charge transfer and/or incorrect spin treatment in the transition region in cases like CO, where there are low-lying π* orbitals and large exchange interactions between the core 1s and valence-acceptor π* orbitals. As an alternative, we demonstrate by comparing with experimental data that a linear response approach using TD-DFT employing common exchange-correlation functionals and finite-size clusters can yield a good description of the spectral evolution of the 1s → π* transition with correct spin and gas-to-chemisorbed chemical shifts in good agreement with experiment.</p>","PeriodicalId":48683,"journal":{"name":"Structural Dynamics-Us","volume":null,"pages":null},"PeriodicalIF":2.3000,"publicationDate":"2022-01-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8759799/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Dynamics-Us","FirstCategoryId":"101","ListUrlMain":"https://doi.org/10.1063/4.0000135","RegionNum":2,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
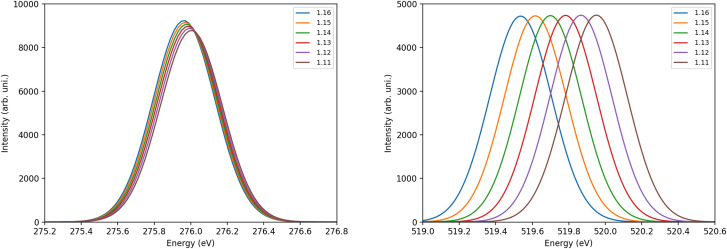
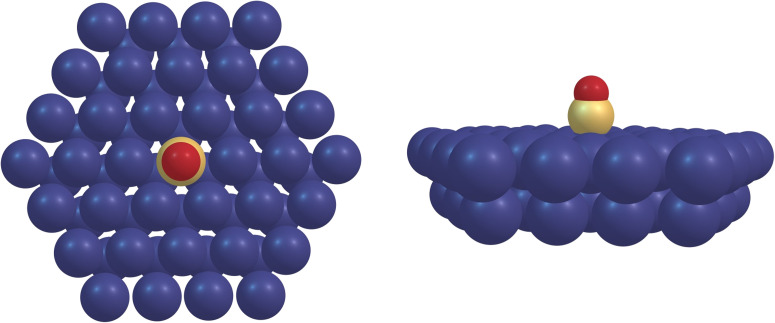
The desorption of a carbon monoxide molecule from a Ru(0001) surface was studied by means of X-ray Absorption Spectra (XAS) computed with Transition Potential (TP-DFT) and Time Dependent (TD-DFT) DFT methods. By unraveling the evolution of the CO electronic structure upon desorption, we observed that at 2.3 Å from the surface, the CO molecule has already predominantly gas-phase character. While C 1s XAS is quite insensitive to changes in the C-O bond length, the O 1s excitation is very sensitive with the π* coming down in energy upon CO bond stretching, which competes with the increase in orbital energy due to the repulsive interaction with the metallic surface. We show in a systematic way that the TP-DFT method can describe the XAS rather well at the endpoints (chemisorbed and gas phase) but is affected by artificial charge transfer and/or incorrect spin treatment in the transition region in cases like CO, where there are low-lying π* orbitals and large exchange interactions between the core 1s and valence-acceptor π* orbitals. As an alternative, we demonstrate by comparing with experimental data that a linear response approach using TD-DFT employing common exchange-correlation functionals and finite-size clusters can yield a good description of the spectral evolution of the 1s → π* transition with correct spin and gas-to-chemisorbed chemical shifts in good agreement with experiment.
用过渡电位和随时间变化的密度泛函理论方法模拟从 Ru(0001) 解吸 CO 的 X 射线吸收光谱。
我们利用过渡势(TP-DFT)和时变(TD-DFT)DFT 方法计算的 X 射线吸收光谱(XAS)研究了一氧化碳分子从 Ru(0001)表面解吸的过程。通过揭示解吸过程中 CO 电子结构的演变,我们观察到在距离表面 2.3 Å 处,CO 分子已经主要具有气相特性。虽然 C 1s XAS 对 C-O 键长度的变化非常不敏感,但 O 1s 激发却非常敏感,在 CO 键伸展时,π* 的能量会下降,这与与金属表面的排斥作用导致的轨道能量增加形成了竞争。我们用系统的方法表明,TP-DFT 方法可以很好地描述端点(化学吸附和气相)的 XAS,但在 CO 等情况下,会受到过渡区人为电荷转移和/或不正确自旋处理的影响,因为在过渡区存在低洼的 π* 轨道以及核心 1s 与价受 π* 轨道之间的大量交换相互作用。作为一种替代方法,我们通过与实验数据的比较证明,利用普通交换相关函数和有限尺寸簇的 TD-DFT 线性响应方法可以很好地描述 1s → π* 转变的光谱演变,其正确的自旋和气体-化合层化学位移与实验结果非常一致。
Structural Dynamics-UsCHEMISTRY, PHYSICALPHYSICS, ATOMIC, MOLECU-PHYSICS, ATOMIC, MOLECULAR & CHEMICAL
CiteScore
5.50
自引率
3.60%
发文量
24
审稿时长
16 weeks
期刊介绍:
Structural Dynamics focuses on the recent developments in experimental and theoretical methods and techniques that allow a visualization of the electronic and geometric structural changes in real time of chemical, biological, and condensed-matter systems. The community of scientists and engineers working on structural dynamics in such diverse systems often use similar instrumentation and methods.
The journal welcomes articles dealing with fundamental problems of electronic and structural dynamics that are tackled by new methods, such as:
Time-resolved X-ray and electron diffraction and scattering,
Coherent diffractive imaging,
Time-resolved X-ray spectroscopies (absorption, emission, resonant inelastic scattering, etc.),
Time-resolved electron energy loss spectroscopy (EELS) and electron microscopy,
Time-resolved photoelectron spectroscopies (UPS, XPS, ARPES, etc.),
Multidimensional spectroscopies in the infrared, the visible and the ultraviolet,
Nonlinear spectroscopies in the VUV, the soft and the hard X-ray domains,
Theory and computational methods and algorithms for the analysis and description of structuraldynamics and their associated experimental signals.
These new methods are enabled by new instrumentation, such as:
X-ray free electron lasers, which provide flux, coherence, and time resolution,
New sources of ultrashort electron pulses,
New sources of ultrashort vacuum ultraviolet (VUV) to hard X-ray pulses, such as high-harmonic generation (HHG) sources or plasma-based sources,
New sources of ultrashort infrared and terahertz (THz) radiation,
New detectors for X-rays and electrons,
New sample handling and delivery schemes,
New computational capabilities.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


