In Hee Kwak, Na Hee Kim, Hyeo-Il Ma, Young Eun Kim
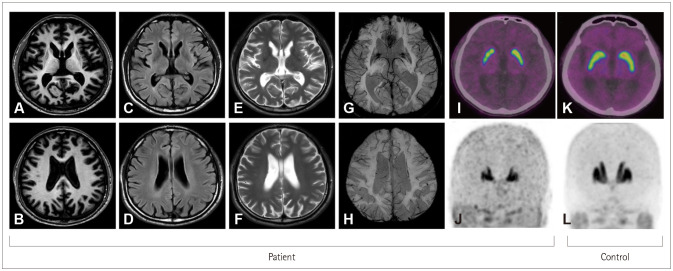
{"title":"Huntington's Disease Presenting as Adult-Onset Parkinsonism.","authors":"In Hee Kwak, Na Hee Kim, Hyeo-Il Ma, Young Eun Kim","doi":"10.3988/jcn.2022.18.1.87","DOIUrl":null,"url":null,"abstract":"Dear Editor, Huntington’s disease (HD) is archetypally characterized as progressive cognitive decline, psychiatric disturbance, and involuntary movements.1-7 Chorea is the most common movement feature, appearing in over 90% of HD patients.3 These symptoms usually start between 30 and 40 years of age, with this variation in the onset age being influenced by the cytosineadenine-guanine (CAG) repeat expansion length.2-4 The Westphal variant of HD that presents as a distinct clinical entity characterized by parkinsonism is prevalent in juvenile-onset HD and has longer CAG expansions than the typical choreic form of adult-onset HD.1-3,7 Therefore, a sole manifestation with parkinsonism in adult-onset HD is rare.1,2,6,8 Previous studies have found that the Westphal variant accounts for 85% of cases of juvenile-onset HD, whereas only 6%–9% of patients with adult-onset HD have initial manifestations of parkinsonism.3,5,6 Here we report the clinical assessment and course of an adult patient with HD who presented as young-onset parkinsonism with a diffuse presynaptic dopaminergic deficit and showed early responsiveness to levodopa. A 34-year-old male developed progressive bradykinesia and gait disturbance over 4 months. He had no underlying medical problems or medication use, but there was a 10-year history of vivid dream and dream enactment behaviors. He was an only child and had no definite family history on his mother’s side. The findings of a neurological examination of his mother were normal. His father died in his 40s after suffering from progressive motor deterioration with psychiatric problems following a traffic collision. No reliable family history was obtained from his father or other relatives because he had not had contact with them since he was a child. A neurological examination of the patient revealed generalized symmetric parkinsonism including masked face, bradykinesia, rigidity, and postural instability (Supplementary Video 1 in the online-only Data Supplement). His speech was mildly dysarthric, and deep tendon reflexes were increased. He had a score of 36 on the Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscale, and was at stage 3 on the Hoehn and Yahr scale. Administering levodopa at 200 mg daily provided symptomatic improvement, and his UPDRS motor score reduced to 26. Possible causes of young-onset parkinsonism were evaluated based on these findings. Brain magnetic resonance imaging (MRI) using various imaging sequences did not reveal any signal abnormality other than brain atrophy (Fig. 1A-H). 18F-FP-CIT positron-emission tomography (PET) for presynaptic dopamine transporter imaging revealed symmetric decreased uptake in the bilateral striatum including caudate and putamen overall compared with a healthy control (Fig. 1I-L). Considering the unclear family history, gene tests were performed for spinocerebellar ataxia 2 and PARK2, and the findings for both were normal. However, unlike for cognition, the scores on the Mini Mental State Examination and Montreal Cognitive Assessment were unexpectedly low at 23 and 22. MRI showed mild diffuse cortical atrophy and caudate atrophy (Fig. 1A-H). An examination of his eye movement revealed delayed saccadic initiation without any gaze limitation. Based on these findings, geIn Hee Kwak Na Hee Kim Hyeo-il Ma Young Eun Kim","PeriodicalId":324902,"journal":{"name":"Journal of Clinical Neurology (Seoul, Korea)","volume":" ","pages":"87-89"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/dd/7e/jcn-18-87.PMC8762510.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Neurology (Seoul, Korea)","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3988/jcn.2022.18.1.87","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
Dear Editor, Huntington’s disease (HD) is archetypally characterized as progressive cognitive decline, psychiatric disturbance, and involuntary movements.1-7 Chorea is the most common movement feature, appearing in over 90% of HD patients.3 These symptoms usually start between 30 and 40 years of age, with this variation in the onset age being influenced by the cytosineadenine-guanine (CAG) repeat expansion length.2-4 The Westphal variant of HD that presents as a distinct clinical entity characterized by parkinsonism is prevalent in juvenile-onset HD and has longer CAG expansions than the typical choreic form of adult-onset HD.1-3,7 Therefore, a sole manifestation with parkinsonism in adult-onset HD is rare.1,2,6,8 Previous studies have found that the Westphal variant accounts for 85% of cases of juvenile-onset HD, whereas only 6%–9% of patients with adult-onset HD have initial manifestations of parkinsonism.3,5,6 Here we report the clinical assessment and course of an adult patient with HD who presented as young-onset parkinsonism with a diffuse presynaptic dopaminergic deficit and showed early responsiveness to levodopa. A 34-year-old male developed progressive bradykinesia and gait disturbance over 4 months. He had no underlying medical problems or medication use, but there was a 10-year history of vivid dream and dream enactment behaviors. He was an only child and had no definite family history on his mother’s side. The findings of a neurological examination of his mother were normal. His father died in his 40s after suffering from progressive motor deterioration with psychiatric problems following a traffic collision. No reliable family history was obtained from his father or other relatives because he had not had contact with them since he was a child. A neurological examination of the patient revealed generalized symmetric parkinsonism including masked face, bradykinesia, rigidity, and postural instability (Supplementary Video 1 in the online-only Data Supplement). His speech was mildly dysarthric, and deep tendon reflexes were increased. He had a score of 36 on the Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscale, and was at stage 3 on the Hoehn and Yahr scale. Administering levodopa at 200 mg daily provided symptomatic improvement, and his UPDRS motor score reduced to 26. Possible causes of young-onset parkinsonism were evaluated based on these findings. Brain magnetic resonance imaging (MRI) using various imaging sequences did not reveal any signal abnormality other than brain atrophy (Fig. 1A-H). 18F-FP-CIT positron-emission tomography (PET) for presynaptic dopamine transporter imaging revealed symmetric decreased uptake in the bilateral striatum including caudate and putamen overall compared with a healthy control (Fig. 1I-L). Considering the unclear family history, gene tests were performed for spinocerebellar ataxia 2 and PARK2, and the findings for both were normal. However, unlike for cognition, the scores on the Mini Mental State Examination and Montreal Cognitive Assessment were unexpectedly low at 23 and 22. MRI showed mild diffuse cortical atrophy and caudate atrophy (Fig. 1A-H). An examination of his eye movement revealed delayed saccadic initiation without any gaze limitation. Based on these findings, geIn Hee Kwak Na Hee Kim Hyeo-il Ma Young Eun Kim

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


