Chaoxi Zhou, Fujun Wang, Hongfang Ma, Na Xing, Lin Hou, Yaping Du, Haixia Ding
{"title":"Silencing of FOS-like antigen 1 represses restenosis <i>via</i> the ERK/AP-1 pathway in type 2 diabetic mice.","authors":"Chaoxi Zhou, Fujun Wang, Hongfang Ma, Na Xing, Lin Hou, Yaping Du, Haixia Ding","doi":"10.1177/14791641211058855","DOIUrl":null,"url":null,"abstract":"<p><p>Restenosis is a major limiting factor for a successful outcome in type 2 diabetes (T2D) patients undergoing percutaneous coronary intervention (PCI). The aim of this study is to explore the role and regulatory mechanism of FOS-like antigen 1 (FOSL1) in restenosis in T2D. A T2D with restenosis mouse model was established by the combination of high-fat diet and streptozotocin injection and by wire-injury. High glucose (HG)-treated vascular smooth muscle cells (VSMCs) were used to mimic T2D in vitro. The results of quantitative real time PCR and western blotting demonstrated that the expression of FOSL1 was increased not only in T2D mice or HG-induced VSMCs, but also in T2D mice that underwent wire-injury. HE staining revealed that FOSL1 knockdown significantly reduced the intimal/media ratio of T2D mice after wire-injury. Silencing of FOSL1 reversed the promoting effects of HG treatment on viability, migration and inflammation reactions, and the inhibiting effect on the apoptosis of VSMCs. Inhibition of ERK/AP-1 pathway obtained similar patterns in HG-induced VSMCs. The activation of ERK/AP-1 pathway reversed the influence of FOSL1 knockdown on HG-induced VSMCs. Our findings indicate that silencing of FOSL1 may suppress restenosis via regulation of the ERK/AP-1 pathway in T2D mice, pointing out a potential therapeutic target to prevent restenosis in T2D.</p>","PeriodicalId":11092,"journal":{"name":"Diabetes & Vascular Disease Research","volume":"18 6","pages":"14791641211058855"},"PeriodicalIF":3.0000,"publicationDate":"2021-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ae/60/10.1177_14791641211058855.PMC8669130.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Diabetes & Vascular Disease Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1177/14791641211058855","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 3
Abstract
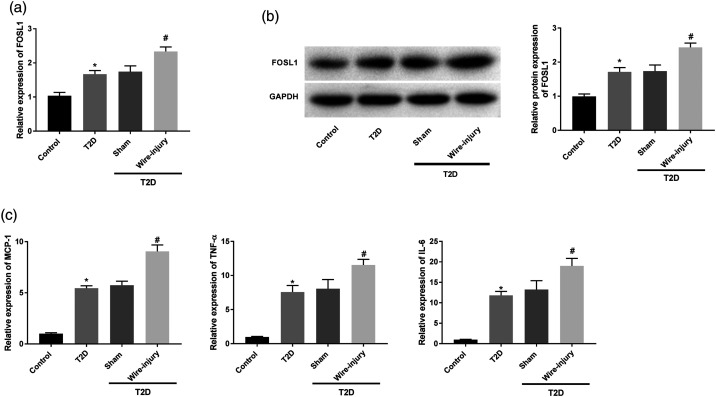
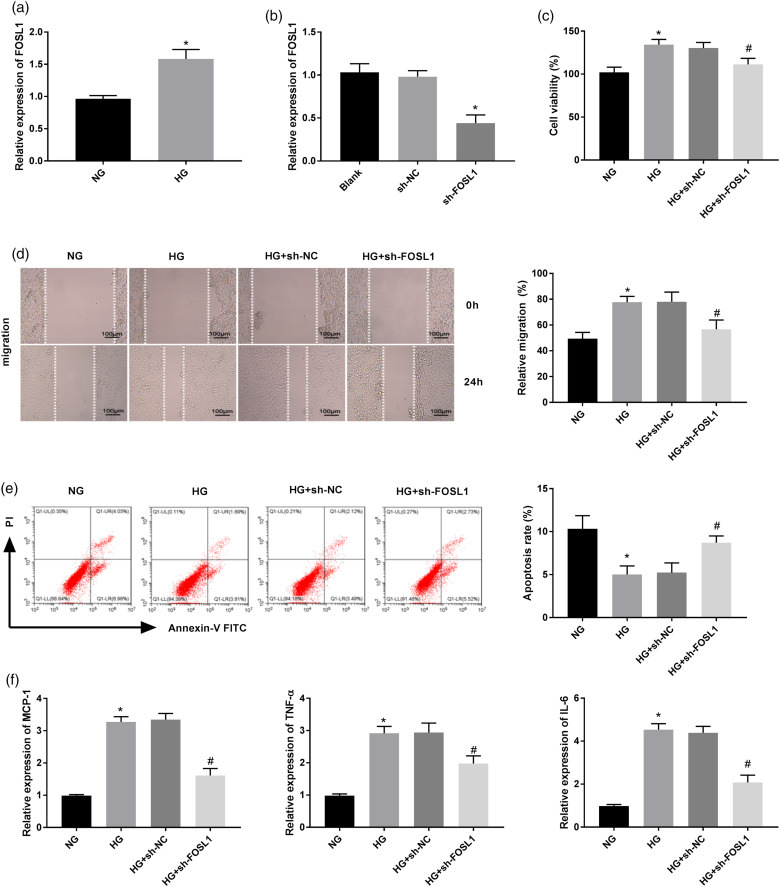
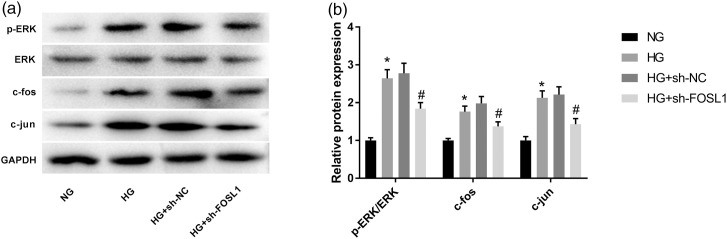
Restenosis is a major limiting factor for a successful outcome in type 2 diabetes (T2D) patients undergoing percutaneous coronary intervention (PCI). The aim of this study is to explore the role and regulatory mechanism of FOS-like antigen 1 (FOSL1) in restenosis in T2D. A T2D with restenosis mouse model was established by the combination of high-fat diet and streptozotocin injection and by wire-injury. High glucose (HG)-treated vascular smooth muscle cells (VSMCs) were used to mimic T2D in vitro. The results of quantitative real time PCR and western blotting demonstrated that the expression of FOSL1 was increased not only in T2D mice or HG-induced VSMCs, but also in T2D mice that underwent wire-injury. HE staining revealed that FOSL1 knockdown significantly reduced the intimal/media ratio of T2D mice after wire-injury. Silencing of FOSL1 reversed the promoting effects of HG treatment on viability, migration and inflammation reactions, and the inhibiting effect on the apoptosis of VSMCs. Inhibition of ERK/AP-1 pathway obtained similar patterns in HG-induced VSMCs. The activation of ERK/AP-1 pathway reversed the influence of FOSL1 knockdown on HG-induced VSMCs. Our findings indicate that silencing of FOSL1 may suppress restenosis via regulation of the ERK/AP-1 pathway in T2D mice, pointing out a potential therapeutic target to prevent restenosis in T2D.
期刊介绍:
Diabetes & Vascular Disease Research is the first international peer-reviewed journal to unite diabetes and vascular disease in a single title. The journal publishes original papers, research letters and reviews. This journal is a member of the Committee on Publication Ethics (COPE)

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


