Jelena De Vrieze, Ingrid M B H van de Laar, Johanneke F de Rijk-van Andel, Erik-Jan Kamsteeg, Irene A W Kotsopoulos, Stella A de Man
{"title":"Expanding Phenotype of <i>ATP1A3</i> - Related Disorders: A Case Series.","authors":"Jelena De Vrieze, Ingrid M B H van de Laar, Johanneke F de Rijk-van Andel, Erik-Jan Kamsteeg, Irene A W Kotsopoulos, Stella A de Man","doi":"10.1177/2329048X211048068","DOIUrl":null,"url":null,"abstract":"<p><p>Neurologic disorders caused by mutations in the <i>ATP1A3</i> gene were originally reported as three distinct rare clinical syndromes: Alternating Hemiplegia of Childhood (AHC), Rapid-onset Dystonia Parkinsonism (RDP) and Cerebellar ataxia, Areflexia, Pes cavus, Opticus atrophy and Sensorineural hearing loss (CAPOS). In this case series, we describe 3 patients. A mother and her daughter showed an intermediate phenotype different from each other with the same heterozygous missense mutation (p.[R756C]), recently described in literature as Relapsing Encephalopathy With Cerebellar Ataxia (RECA). In addition, a third patient showed an intermediate AHC-RDP phenotype and had a likely pathogenic novel de novo missense mutation (p.[L100 V]). These patients support the growing evidence that AHC, RDP and RECA are part of a continuous <i>ATP1A3</i> mutation spectrum that is still expanding. Three common features were a sudden onset, asymmetrical neurological symptoms, as well as the presence of triggering factors. When present, the authors argue to perform exome sequencing in an early stage.</p>","PeriodicalId":72572,"journal":{"name":"Child neurology open","volume":"8 ","pages":"2329048X211048068"},"PeriodicalIF":0.0000,"publicationDate":"2021-11-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/62/f1/10.1177_2329048X211048068.PMC8573619.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Child neurology open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2329048X211048068","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
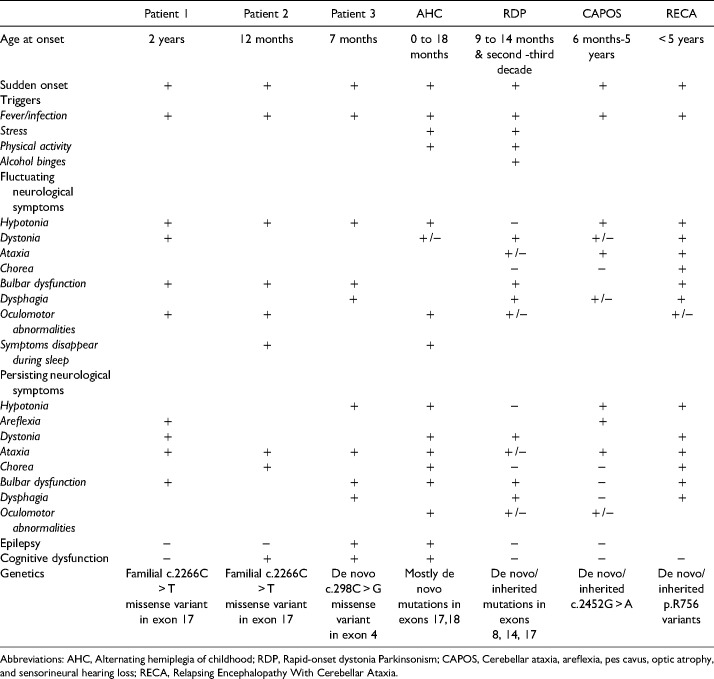
Neurologic disorders caused by mutations in the ATP1A3 gene were originally reported as three distinct rare clinical syndromes: Alternating Hemiplegia of Childhood (AHC), Rapid-onset Dystonia Parkinsonism (RDP) and Cerebellar ataxia, Areflexia, Pes cavus, Opticus atrophy and Sensorineural hearing loss (CAPOS). In this case series, we describe 3 patients. A mother and her daughter showed an intermediate phenotype different from each other with the same heterozygous missense mutation (p.[R756C]), recently described in literature as Relapsing Encephalopathy With Cerebellar Ataxia (RECA). In addition, a third patient showed an intermediate AHC-RDP phenotype and had a likely pathogenic novel de novo missense mutation (p.[L100 V]). These patients support the growing evidence that AHC, RDP and RECA are part of a continuous ATP1A3 mutation spectrum that is still expanding. Three common features were a sudden onset, asymmetrical neurological symptoms, as well as the presence of triggering factors. When present, the authors argue to perform exome sequencing in an early stage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


