Microsecond molecular dynamics suggest that a non-synonymous mutation, frequently observed in patients with mild symptoms in Tokyo, alters dynamics of the SARS-CoV-2 main protease.
{"title":"Microsecond molecular dynamics suggest that a non-synonymous mutation, frequently observed in patients with mild symptoms in Tokyo, alters dynamics of the SARS-CoV-2 main protease.","authors":"Daisuke Kuroda, Kouhei Tsumoto","doi":"10.2142/biophysico.bppb-v18.022","DOIUrl":null,"url":null,"abstract":"<p><p>Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the coronavirus disease 2019 (COVID-19), spread rapidly around the globe. The main protease encoded by SARS-CoV-2 is essential for processing of the polyproteins translated from the viral RNA genome, making this protein a potential drug target. A recently reported mutation in the protease, P108S, may be responsible for milder symptoms observed in COVID-19 patients in Tokyo. Starting from a crystal structure of the SARS-CoV-2 main protease in the dimeric form, we performed triplicate 5.0-μs molecular dynamics simulations of the wild-type and P108S mutant. Our computational results suggest a link between the mutation P108S and dynamics of the catalytic sites in the main protease: The catalytic dyad become considerably inaccessible to substrates in the P108S mutant. Our results also demonstrate the potential of molecular dynamics simulations to complement experimental techniques and other computational methods, such as protein design calculations, which predict effects of mutations based on static crystal structures. Further studies are certainly necessary to quantitively understand the relationships between the P108S mutation and physical properties of the main protease, but the results of our study will immediately inform development of new protease inhibitors.</p>","PeriodicalId":8976,"journal":{"name":"Biophysics and Physicobiology","volume":" ","pages":"215-222"},"PeriodicalIF":0.0000,"publicationDate":"2021-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f7/18/18_215.PMC8470906.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysics and Physicobiology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2142/biophysico.bppb-v18.022","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
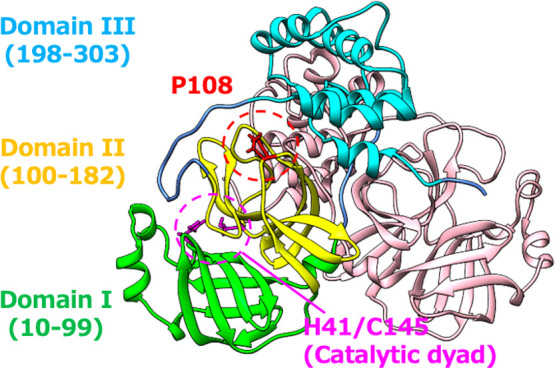
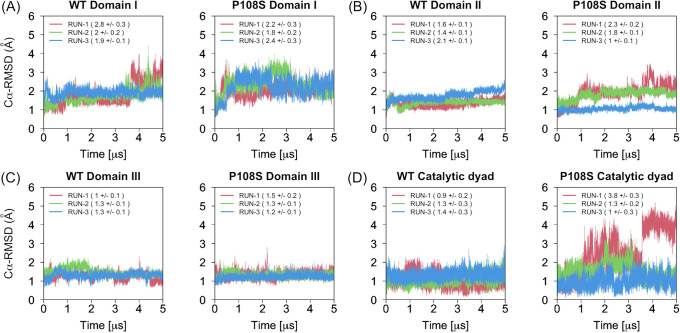
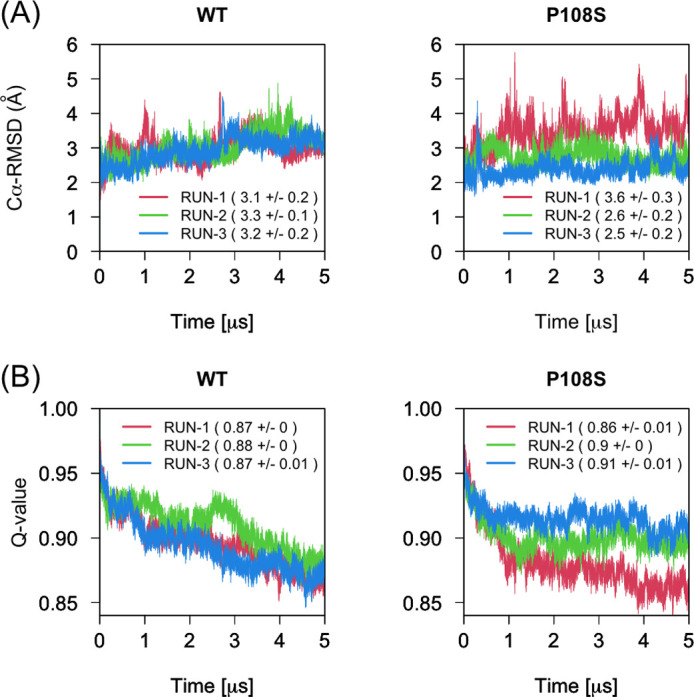
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the coronavirus disease 2019 (COVID-19), spread rapidly around the globe. The main protease encoded by SARS-CoV-2 is essential for processing of the polyproteins translated from the viral RNA genome, making this protein a potential drug target. A recently reported mutation in the protease, P108S, may be responsible for milder symptoms observed in COVID-19 patients in Tokyo. Starting from a crystal structure of the SARS-CoV-2 main protease in the dimeric form, we performed triplicate 5.0-μs molecular dynamics simulations of the wild-type and P108S mutant. Our computational results suggest a link between the mutation P108S and dynamics of the catalytic sites in the main protease: The catalytic dyad become considerably inaccessible to substrates in the P108S mutant. Our results also demonstrate the potential of molecular dynamics simulations to complement experimental techniques and other computational methods, such as protein design calculations, which predict effects of mutations based on static crystal structures. Further studies are certainly necessary to quantitively understand the relationships between the P108S mutation and physical properties of the main protease, but the results of our study will immediately inform development of new protease inhibitors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



