Hooman Zabeti, Nick Dexter, Amir Hosein Safari, Nafiseh Sedaghat, Maxwell Libbrecht, Leonid Chindelevitch
{"title":"INGOT-DR: an interpretable classifier for predicting drug resistance in M. tuberculosis.","authors":"Hooman Zabeti, Nick Dexter, Amir Hosein Safari, Nafiseh Sedaghat, Maxwell Libbrecht, Leonid Chindelevitch","doi":"10.1186/s13015-021-00198-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Prediction of drug resistance and identification of its mechanisms in bacteria such as Mycobacterium tuberculosis, the etiological agent of tuberculosis, is a challenging problem. Solving this problem requires a transparent, accurate, and flexible predictive model. The methods currently used for this purpose rarely satisfy all of these criteria. On the one hand, approaches based on testing strains against a catalogue of previously identified mutations often yield poor predictive performance; on the other hand, machine learning techniques typically have higher predictive accuracy, but often lack interpretability and may learn patterns that produce accurate predictions for the wrong reasons. Current interpretable methods may either exhibit a lower accuracy or lack the flexibility needed to generalize them to previously unseen data.</p><p><strong>Contribution: </strong>In this paper we propose a novel technique, inspired by group testing and Boolean compressed sensing, which yields highly accurate predictions, interpretable results, and is flexible enough to be optimized for various evaluation metrics at the same time.</p><p><strong>Results: </strong>We test the predictive accuracy of our approach on five first-line and seven second-line antibiotics used for treating tuberculosis. We find that it has a higher or comparable accuracy to that of commonly used machine learning models, and is able to identify variants in genes with previously reported association to drug resistance. Our method is intrinsically interpretable, and can be customized for different evaluation metrics. Our implementation is available at github.com/hoomanzabeti/INGOT_DR and can be installed via The Python Package Index (Pypi) under ingotdr. This package is also compatible with most of the tools in the Scikit-learn machine learning library.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"16 1","pages":"17"},"PeriodicalIF":1.7000,"publicationDate":"2021-08-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8353837/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-021-00198-1","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Prediction of drug resistance and identification of its mechanisms in bacteria such as Mycobacterium tuberculosis, the etiological agent of tuberculosis, is a challenging problem. Solving this problem requires a transparent, accurate, and flexible predictive model. The methods currently used for this purpose rarely satisfy all of these criteria. On the one hand, approaches based on testing strains against a catalogue of previously identified mutations often yield poor predictive performance; on the other hand, machine learning techniques typically have higher predictive accuracy, but often lack interpretability and may learn patterns that produce accurate predictions for the wrong reasons. Current interpretable methods may either exhibit a lower accuracy or lack the flexibility needed to generalize them to previously unseen data.
Contribution: In this paper we propose a novel technique, inspired by group testing and Boolean compressed sensing, which yields highly accurate predictions, interpretable results, and is flexible enough to be optimized for various evaluation metrics at the same time.
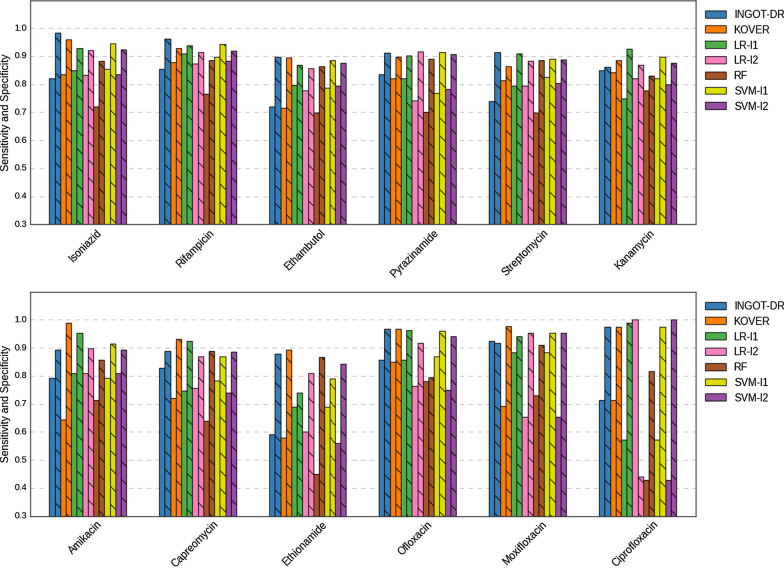
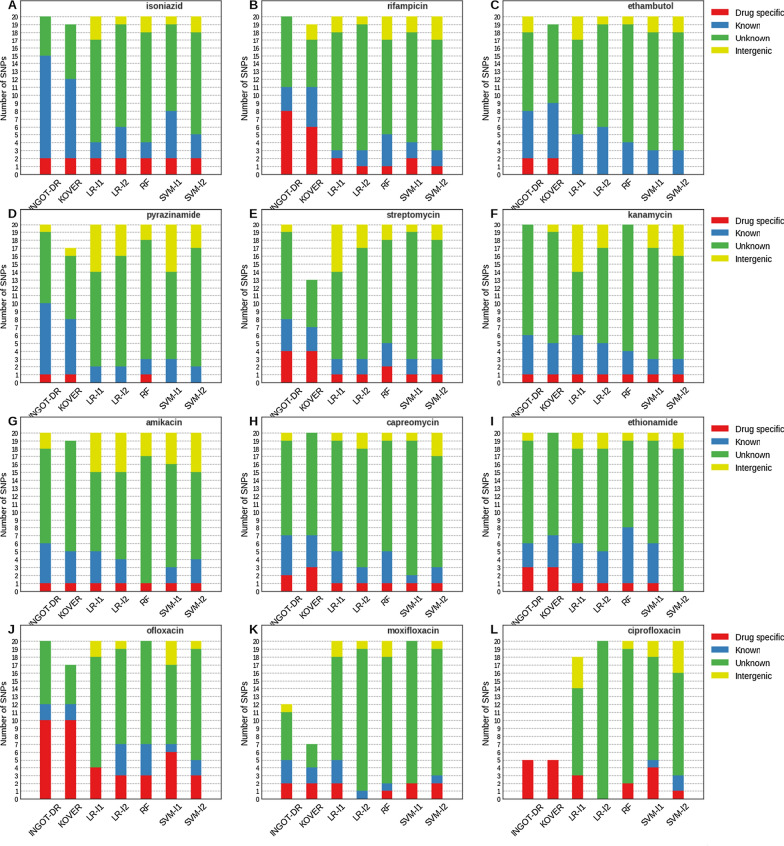
Results: We test the predictive accuracy of our approach on five first-line and seven second-line antibiotics used for treating tuberculosis. We find that it has a higher or comparable accuracy to that of commonly used machine learning models, and is able to identify variants in genes with previously reported association to drug resistance. Our method is intrinsically interpretable, and can be customized for different evaluation metrics. Our implementation is available at github.com/hoomanzabeti/INGOT_DR and can be installed via The Python Package Index (Pypi) under ingotdr. This package is also compatible with most of the tools in the Scikit-learn machine learning library.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


