Sven Schrinner, Manish Goel, Michael Wulfert, Philipp Spohr, Korbinian Schneeberger, Gunnar W Klau
{"title":"Using the longest run subsequence problem within homology-based scaffolding.","authors":"Sven Schrinner, Manish Goel, Michael Wulfert, Philipp Spohr, Korbinian Schneeberger, Gunnar W Klau","doi":"10.1186/s13015-021-00191-8","DOIUrl":null,"url":null,"abstract":"<p><p>Genome assembly is one of the most important problems in computational genomics. Here, we suggest addressing an issue that arises in homology-based scaffolding, that is, when linking and ordering contigs to obtain larger pseudo-chromosomes by means of a second incomplete assembly of a related species. The idea is to use alignments of binned regions in one contig to find the most homologous contig in the other assembly. We show that ordering the contigs of the other assembly can be expressed by a new string problem, the longest run subsequence problem (LRS). We show that LRS is NP-hard and present reduction rules and two algorithmic approaches that, together, are able to solve large instances of LRS to provable optimality. All data used in the experiments as well as our source code are freely available. We demonstrate its usefulness within an existing larger scaffolding approach by solving realistic instances resulting from partial Arabidopsis thaliana assemblies in short computation time.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"16 1","pages":"11"},"PeriodicalIF":1.5000,"publicationDate":"2021-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8240273/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-021-00191-8","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 2
Abstract
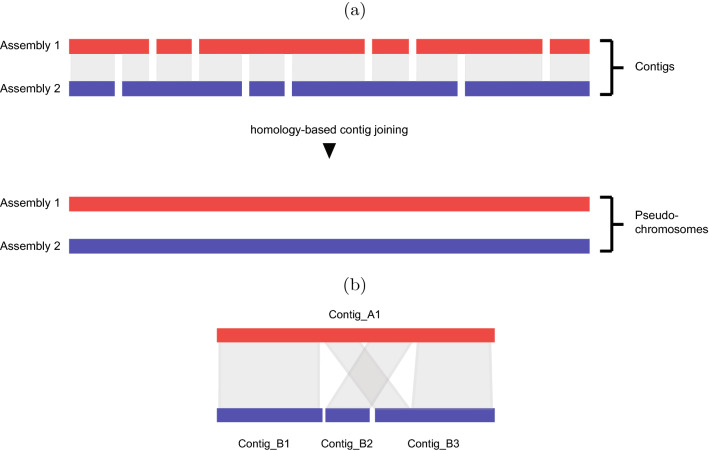
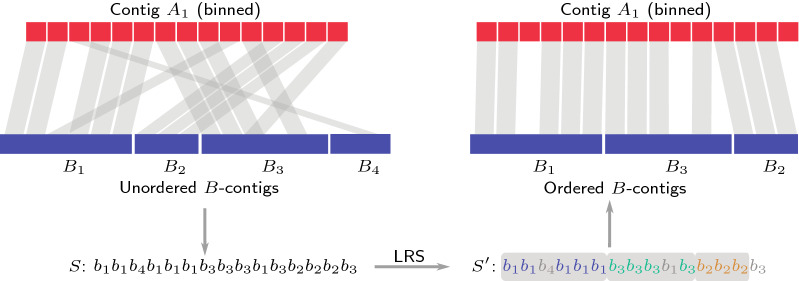
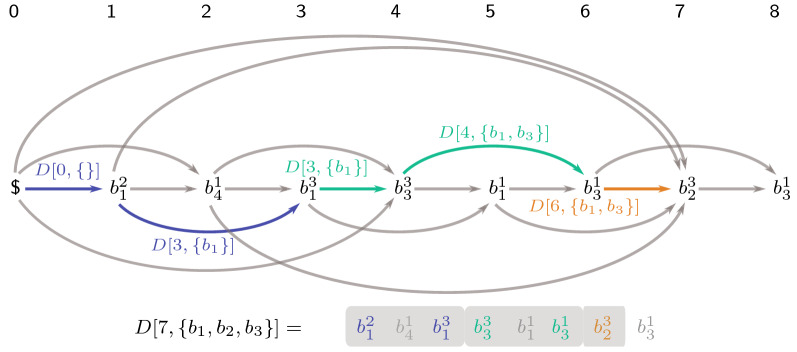
Genome assembly is one of the most important problems in computational genomics. Here, we suggest addressing an issue that arises in homology-based scaffolding, that is, when linking and ordering contigs to obtain larger pseudo-chromosomes by means of a second incomplete assembly of a related species. The idea is to use alignments of binned regions in one contig to find the most homologous contig in the other assembly. We show that ordering the contigs of the other assembly can be expressed by a new string problem, the longest run subsequence problem (LRS). We show that LRS is NP-hard and present reduction rules and two algorithmic approaches that, together, are able to solve large instances of LRS to provable optimality. All data used in the experiments as well as our source code are freely available. We demonstrate its usefulness within an existing larger scaffolding approach by solving realistic instances resulting from partial Arabidopsis thaliana assemblies in short computation time.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


