{"title":"Exact transcript quantification over splice graphs.","authors":"Cong Ma, Hongyu Zheng, Carl Kingsford","doi":"10.1186/s13015-021-00184-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The probability of sequencing a set of RNA-seq reads can be directly modeled using the abundances of splice junctions in splice graphs instead of the abundances of a list of transcripts. We call this model graph quantification, which was first proposed by Bernard et al. (Bioinformatics 30:2447-55, 2014). The model can be viewed as a generalization of transcript expression quantification where every full path in the splice graph is a possible transcript. However, the previous graph quantification model assumes the length of single-end reads or paired-end fragments is fixed.</p><p><strong>Results: </strong>We provide an improvement of this model to handle variable-length reads or fragments and incorporate bias correction. We prove that our model is equivalent to running a transcript quantifier with exactly the set of all compatible transcripts. The key to our method is constructing an extension of the splice graph based on Aho-Corasick automata. The proof of equivalence is based on a novel reparameterization of the read generation model of a state-of-art transcript quantification method.</p><p><strong>Conclusion: </strong>We propose a new approach for graph quantification, which is useful for modeling scenarios where reference transcriptome is incomplete or not available and can be further used in transcriptome assembly or alternative splicing analysis.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"16 1","pages":"5"},"PeriodicalIF":1.7000,"publicationDate":"2021-05-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-021-00184-7","citationCount":"10","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-021-00184-7","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 10
Abstract
Background: The probability of sequencing a set of RNA-seq reads can be directly modeled using the abundances of splice junctions in splice graphs instead of the abundances of a list of transcripts. We call this model graph quantification, which was first proposed by Bernard et al. (Bioinformatics 30:2447-55, 2014). The model can be viewed as a generalization of transcript expression quantification where every full path in the splice graph is a possible transcript. However, the previous graph quantification model assumes the length of single-end reads or paired-end fragments is fixed.
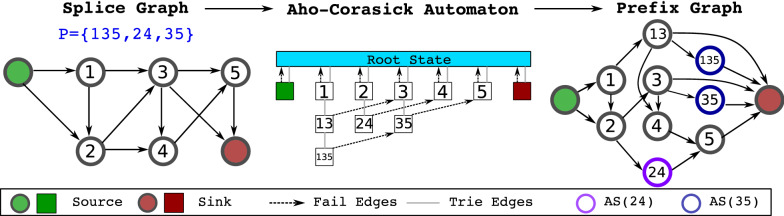
Results: We provide an improvement of this model to handle variable-length reads or fragments and incorporate bias correction. We prove that our model is equivalent to running a transcript quantifier with exactly the set of all compatible transcripts. The key to our method is constructing an extension of the splice graph based on Aho-Corasick automata. The proof of equivalence is based on a novel reparameterization of the read generation model of a state-of-art transcript quantification method.
Conclusion: We propose a new approach for graph quantification, which is useful for modeling scenarios where reference transcriptome is incomplete or not available and can be further used in transcriptome assembly or alternative splicing analysis.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


