{"title":"Developing a Kinase-Specific Target Selection Method Using a Structure-Based Machine Learning Approach.","authors":"Arina Afanasyeva, Chioko Nagao, Kenji Mizuguchi","doi":"10.2147/AABC.S278900","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Despite recent advances in the drug discovery field, developing selective kinase inhibitors remains a complicated issue for a number of reasons, one of which is that there are striking structural similarities in the ATP-binding pockets of kinases.</p><p><strong>Objective: </strong>To address this problem, we have designed a machine learning model utilizing various structure-based and energy-based descriptors to better characterize protein-ligand interactions.</p><p><strong>Methods: </strong>In this work, we use a dataset of 104 human kinases with available PDB structures and experimental activity data against 1202 small-molecule compounds from the PubChem BioAssay dataset \"Navigating the Kinome\". We propose structure-based interaction descriptors to build activity predicting machine learning model.</p><p><strong>Results and discussion: </strong>We report a ligand-oriented computational method for accurate kinase target prioritizing. Our method shows high accuracy compared to similar structure-based activity prediction methods, and more importantly shows the same prediction accuracy when tested on the special set of structurally remote compounds, showing that it is unbiased to ligand structural similarity in the training set data. We hope that our approach will be useful for the development of novel highly selective kinase inhibitors.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":"13 ","pages":"27-40"},"PeriodicalIF":0.0000,"publicationDate":"2020-12-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/AABC.S278900","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AABC.S278900","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 2
Abstract
Introduction: Despite recent advances in the drug discovery field, developing selective kinase inhibitors remains a complicated issue for a number of reasons, one of which is that there are striking structural similarities in the ATP-binding pockets of kinases.
Objective: To address this problem, we have designed a machine learning model utilizing various structure-based and energy-based descriptors to better characterize protein-ligand interactions.
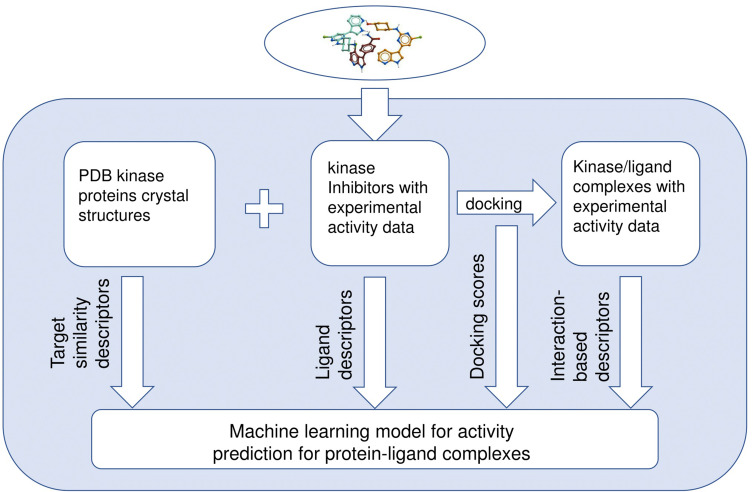
Methods: In this work, we use a dataset of 104 human kinases with available PDB structures and experimental activity data against 1202 small-molecule compounds from the PubChem BioAssay dataset "Navigating the Kinome". We propose structure-based interaction descriptors to build activity predicting machine learning model.
Results and discussion: We report a ligand-oriented computational method for accurate kinase target prioritizing. Our method shows high accuracy compared to similar structure-based activity prediction methods, and more importantly shows the same prediction accuracy when tested on the special set of structurally remote compounds, showing that it is unbiased to ligand structural similarity in the training set data. We hope that our approach will be useful for the development of novel highly selective kinase inhibitors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


