{"title":"An efficient method to transcription factor binding sites imputation via simultaneous completion of multiple matrices with positional consistency†","authors":"Wei-Li Guo and De-Shuang Huang","doi":"10.1039/C7MB00155J","DOIUrl":null,"url":null,"abstract":"<p >Transcription factors (TFs) are DNA-binding proteins that have a central role in regulating gene expression. Identification of DNA-binding sites of TFs is a key task in understanding transcriptional regulation, cellular processes and disease. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) enables genome-wide identification of <em>in vivo</em> TF binding sites. However, it is still difficult to map every TF in every cell line owing to cost and biological material availability, which poses an enormous obstacle for integrated analysis of gene regulation. To address this problem, we propose a novel computational approach, TFBSImpute, for predicting additional TF binding profiles by leveraging information from available ChIP-seq TF binding data. TFBSImpute fuses the dataset to a 3-mode tensor and imputes missing TF binding signals <em>via</em> simultaneous completion of multiple TF binding matrices with positional consistency. We show that signals predicted by our method achieve overall similarity with experimental data and that TFBSImpute significantly outperforms baseline approaches, by assessing the performance of imputation methods against observed ChIP-seq TF binding profiles. Besides, motif analysis shows that TFBSImpute preforms better in capturing binding motifs enriched in observed data compared with baselines, indicating that the higher performance of TFBSImpute is not simply due to averaging related samples. We anticipate that our approach will constitute a useful complement to experimental mapping of TF binding, which is beneficial for further study of regulation mechanisms and disease.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 9","pages":" 1827-1837"},"PeriodicalIF":3.7430,"publicationDate":"2017-07-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C7MB00155J","citationCount":"22","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c7mb00155j","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 22
Abstract
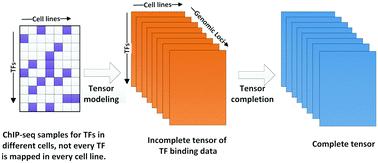
Transcription factors (TFs) are DNA-binding proteins that have a central role in regulating gene expression. Identification of DNA-binding sites of TFs is a key task in understanding transcriptional regulation, cellular processes and disease. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) enables genome-wide identification of in vivo TF binding sites. However, it is still difficult to map every TF in every cell line owing to cost and biological material availability, which poses an enormous obstacle for integrated analysis of gene regulation. To address this problem, we propose a novel computational approach, TFBSImpute, for predicting additional TF binding profiles by leveraging information from available ChIP-seq TF binding data. TFBSImpute fuses the dataset to a 3-mode tensor and imputes missing TF binding signals via simultaneous completion of multiple TF binding matrices with positional consistency. We show that signals predicted by our method achieve overall similarity with experimental data and that TFBSImpute significantly outperforms baseline approaches, by assessing the performance of imputation methods against observed ChIP-seq TF binding profiles. Besides, motif analysis shows that TFBSImpute preforms better in capturing binding motifs enriched in observed data compared with baselines, indicating that the higher performance of TFBSImpute is not simply due to averaging related samples. We anticipate that our approach will constitute a useful complement to experimental mapping of TF binding, which is beneficial for further study of regulation mechanisms and disease.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


