Maria Santa Rocca, Gioia Piatti, Angela Michelucci, Raffaella Guazzo, Veronica Bertini, Cinzia Vinanzi, Maria Adelaide Caligo, Angelo Valetto, Carlo Foresta
{"title":"A novel genetic variant in DNAI2 detected by custom gene panel in a newborn with Primary Ciliary Dyskinesia: case report.","authors":"Maria Santa Rocca, Gioia Piatti, Angela Michelucci, Raffaella Guazzo, Veronica Bertini, Cinzia Vinanzi, Maria Adelaide Caligo, Angelo Valetto, Carlo Foresta","doi":"10.1186/s12881-020-01160-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary ciliary dyskinesia (PCD) is a highly heterogeneous genetic disorder caused by defects in motile cilia. The hallmark features of PCD are the chronic infections of the respiratory tract, moreover, clinical manifestations include also laterality defects and risk of male infertility. Clinical phenotypes of PCD are the result of mutations in genes encoding components of axonema or factors involved in axonemal assembly. Recent studies have identified over 45 PCD-associated genes, therefore, molecular analysis represents a powerful diagnostic tool to confirm and uncover new genetic causes of this rare disease.</p><p><strong>Case presentation: </strong>Here, we describe a female infant of Moroccan origin with normal pressure hydrocephalus (NPH) in addition to most common PCD symptoms. Transmission Electron Microscopy (TEM) and molecular tests, such as a Next generation Sequencing panel and a custom array CGH, were performed for diagnosis of PCD. TEM revealed outer dynein arm (ODA) defects, whilst molecular analyses detected a novel 6,9 kb microdeletion in DNAI2 gene.</p><p><strong>Conclusions: </strong>Since DNAI2 mutations are very rare, this case report contributes to better delineate the important role of DNAI2 as causative of PCD phenotype, suggesting, furthermore, that the variations in DNAI2 may be as a new genetic risk factor for NPH. Indeed, although the association of hydrocephalus with PCD has been well documented, however, only a small number of human patients show this defect. Furthermore, this study highlights the importance of high-throughput technologies in advancing our understanding of heterogeneous genetic disorders.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"220"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01160-5","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01160-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 5
Abstract
Background: Primary ciliary dyskinesia (PCD) is a highly heterogeneous genetic disorder caused by defects in motile cilia. The hallmark features of PCD are the chronic infections of the respiratory tract, moreover, clinical manifestations include also laterality defects and risk of male infertility. Clinical phenotypes of PCD are the result of mutations in genes encoding components of axonema or factors involved in axonemal assembly. Recent studies have identified over 45 PCD-associated genes, therefore, molecular analysis represents a powerful diagnostic tool to confirm and uncover new genetic causes of this rare disease.
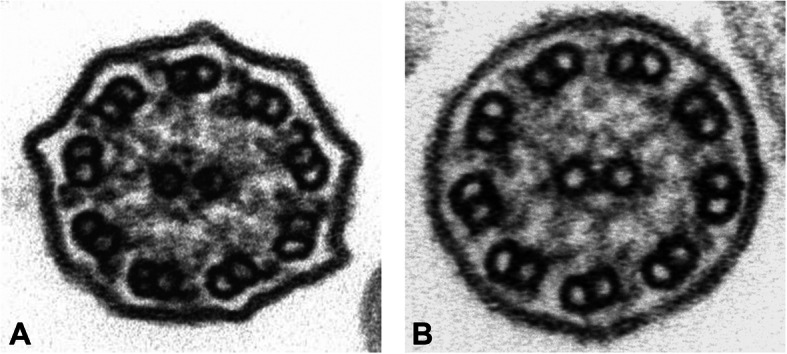
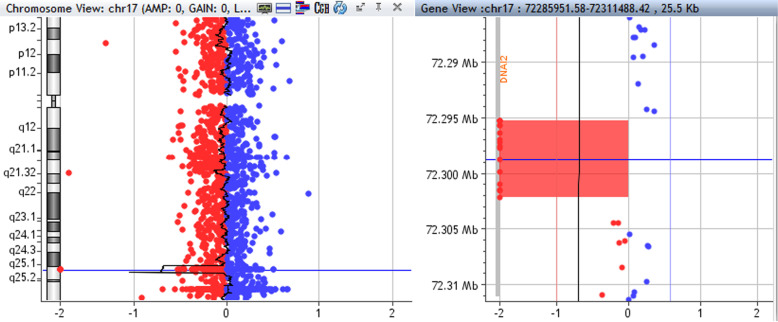
Case presentation: Here, we describe a female infant of Moroccan origin with normal pressure hydrocephalus (NPH) in addition to most common PCD symptoms. Transmission Electron Microscopy (TEM) and molecular tests, such as a Next generation Sequencing panel and a custom array CGH, were performed for diagnosis of PCD. TEM revealed outer dynein arm (ODA) defects, whilst molecular analyses detected a novel 6,9 kb microdeletion in DNAI2 gene.
Conclusions: Since DNAI2 mutations are very rare, this case report contributes to better delineate the important role of DNAI2 as causative of PCD phenotype, suggesting, furthermore, that the variations in DNAI2 may be as a new genetic risk factor for NPH. Indeed, although the association of hydrocephalus with PCD has been well documented, however, only a small number of human patients show this defect. Furthermore, this study highlights the importance of high-throughput technologies in advancing our understanding of heterogeneous genetic disorders.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


