Yuping Niu, Sexin Huang, Zeyu Wang, Peiwen Xu, Lijuan Wang, Jie Li, Ming Gao, Xuan Gao, Yuan Gao
{"title":"A nonsense variant in FBN1 caused autosomal dominant Marfan syndrome in a Chinese family: a case report.","authors":"Yuping Niu, Sexin Huang, Zeyu Wang, Peiwen Xu, Lijuan Wang, Jie Li, Ming Gao, Xuan Gao, Yuan Gao","doi":"10.1186/s12881-020-01148-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Marfan syndrome (MFS) is a common autosomal dominant inherited disease, and the occurrence rate is around 0.1-0.2‰. The causative variant of FNB1 gene accounts for approximately 70-80% of all MFS cases. In this study, we found a heterozygous c.3217G > T (p.Glu1073*) nonsense variant in the FBN1 gene. This finding extended the variant spectrum of the FBN1 gene and will provide a solution for patients to bear healthy offspring by preimplantation genetic testing or prenatal diagnosis.</p><p><strong>Case presentation: </strong>The patient was treated due to tachycardia during excitement in a hospital. Echocardiography showed dilatation of the ascending aorta and main pulmonary artery, mitral regurgitation (mild), tricuspid regurgitation (mild), and abnormal left ventricular filling. Electrocardiograph showed sinus rhythm. In addition, flutters of shadows in front of his eyes and vitreous opacity were present in the patient. Genomic DNA was extracted from peripheral blood samples from members of the family and 100 unrelated controls. Potential variants were screened out by next-generation sequencing and confirmed by MLPA & Sanger sequencing. Real-time fluorescence quantitative PCR (RT-qPCR) was performed to detect the relative mRNA quantitation in the patient. A heterozygous nonsense variant c.3217G > T of the FBN1 gene, which resulted in p. Glu1073Term, was identified in both patients. Only wild type bases were found in the cDNA sequence of the patient. Real-time fluorogenic quantitative PCR results showed that the relative expression level of FBN1 cDNA in the patient was only about 21% compared to that of normal individuals. This variant c.3217G > T of the FBN1 gene introduces a Stop codon in the cb-EGF12 domain. We speculated that a premature translational-termination codon (PTC) was located in the mRNA and the target mRNA was disintegrated through a process known as nonsense-mediated mRNA decay (NMD), which led to a significant decrease of the fibrillin-1 protein, eventually causing clinical symptoms in the patient.</p><p><strong>Conclusions: </strong>In this study, we found a heterozygous c.3217G > T (p.Glu1073*) nonsense variant in the FBN1 gene, which eventually led to Marfan syndrome in a Chinese family.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"211"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01148-1","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01148-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 3
Abstract
Background: Marfan syndrome (MFS) is a common autosomal dominant inherited disease, and the occurrence rate is around 0.1-0.2‰. The causative variant of FNB1 gene accounts for approximately 70-80% of all MFS cases. In this study, we found a heterozygous c.3217G > T (p.Glu1073*) nonsense variant in the FBN1 gene. This finding extended the variant spectrum of the FBN1 gene and will provide a solution for patients to bear healthy offspring by preimplantation genetic testing or prenatal diagnosis.
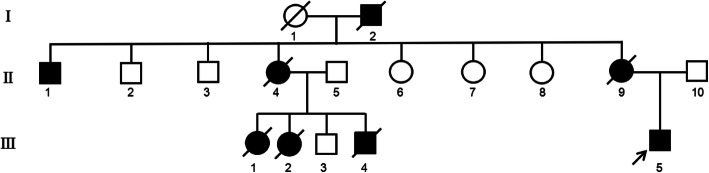
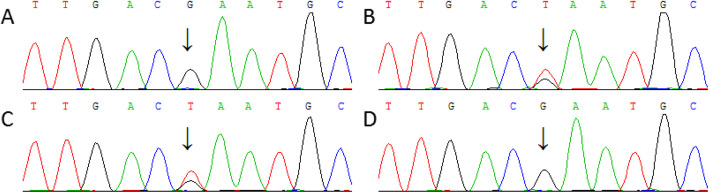
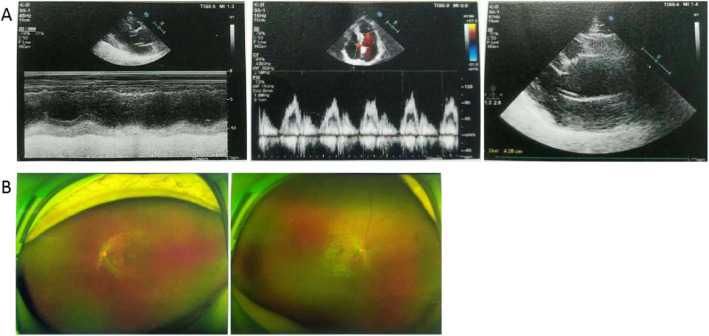
Case presentation: The patient was treated due to tachycardia during excitement in a hospital. Echocardiography showed dilatation of the ascending aorta and main pulmonary artery, mitral regurgitation (mild), tricuspid regurgitation (mild), and abnormal left ventricular filling. Electrocardiograph showed sinus rhythm. In addition, flutters of shadows in front of his eyes and vitreous opacity were present in the patient. Genomic DNA was extracted from peripheral blood samples from members of the family and 100 unrelated controls. Potential variants were screened out by next-generation sequencing and confirmed by MLPA & Sanger sequencing. Real-time fluorescence quantitative PCR (RT-qPCR) was performed to detect the relative mRNA quantitation in the patient. A heterozygous nonsense variant c.3217G > T of the FBN1 gene, which resulted in p. Glu1073Term, was identified in both patients. Only wild type bases were found in the cDNA sequence of the patient. Real-time fluorogenic quantitative PCR results showed that the relative expression level of FBN1 cDNA in the patient was only about 21% compared to that of normal individuals. This variant c.3217G > T of the FBN1 gene introduces a Stop codon in the cb-EGF12 domain. We speculated that a premature translational-termination codon (PTC) was located in the mRNA and the target mRNA was disintegrated through a process known as nonsense-mediated mRNA decay (NMD), which led to a significant decrease of the fibrillin-1 protein, eventually causing clinical symptoms in the patient.
Conclusions: In this study, we found a heterozygous c.3217G > T (p.Glu1073*) nonsense variant in the FBN1 gene, which eventually led to Marfan syndrome in a Chinese family.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


