Histone Purification Combined with High-Resolution Mass Spectrometry to Examine Histone Post-Translational Modifications and Histone Variants in Caenorhabditis elegans
Q1 Biochemistry, Genetics and Molecular Biology
引用次数: 4
Abstract
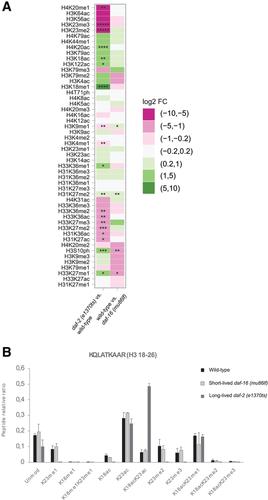
Histones are the major proteinaceous component of chromatin in eukaryotic cells and an important part of the epigenome, affecting most DNA‐related events, including transcription, DNA replication, and chromosome segregation. The properties of histones are greatly influenced by their post‐translational modifications (PTMs), over 200 of which are known today. Given this large number, researchers need sophisticated methods to study histone PTMs comprehensively. In particular, mass spectrometry (MS)−based approaches have gained popularity, allowing for the quantification of dozens of histone PTMs at once. Using these approaches, even the study of co‐occurring PTMs and the discovery of novel PTMs become feasible. The success of MS‐based approaches relies substantially on obtaining pure and well‐preserved histones for analysis, which can be difficult depending on the source material. Caenorhabditis elegans has been a popular model organism to study the epigenome, but isolation of pure histones from these animals has been challenging. Here, we address this issue, presenting a method for efficient isolation of pure histone proteins from C. elegans at good yield. Further, we describe an MS pipeline optimized for accurate relative quantification of histone PTMs from C. elegans. We alkylate and tryptically digest the histones, analyze them by bottom‐up MS, and then evaluate the resulting data by a C. elegans−adapted version of the software EpiProfile 2.0. Finally, we show the utility of this pipeline by determining differences in histone PTMs between C. elegans strains that age at different rates and thereby achieve very different lifespans. © 2020 The Authors.
组蛋白纯化结合高分辨率质谱法检测秀丽隐杆线虫组蛋白翻译后修饰和组蛋白变异
组蛋白是真核细胞中染色质的主要蛋白质成分,也是表观基因组的重要组成部分,影响大多数DNA相关事件,包括转录、DNA复制和染色体分离。组蛋白的特性很大程度上受其翻译后修饰(PTMs)的影响,目前已知的PTMs有200多种。由于数量庞大,研究人员需要复杂的方法来全面研究组蛋白ptm。特别是,基于质谱(MS)的方法已经得到普及,允许一次定量几十个组蛋白PTMs。使用这些方法,甚至研究共同发生的ptm和发现新的ptm也变得可行。基于质谱的方法的成功主要依赖于获得纯的和保存良好的组蛋白进行分析,这可能是困难的,取决于源材料。秀丽隐杆线虫一直是研究表观基因组的流行模式生物,但从这些动物中分离纯组蛋白一直具有挑战性。在这里,我们解决了这个问题,提出了一种从秀丽隐杆线虫中高效分离纯组蛋白的方法。此外,我们描述了一个MS管道,优化了秀丽隐杆线虫组蛋白PTMs的准确相对定量。我们对组蛋白进行烷基化和胰消化,用自下而上的质谱法对其进行分析,然后用适合线虫的EpiProfile 2.0软件对结果数据进行评估。最后,我们通过确定以不同速度衰老的秀丽隐杆线虫菌株之间组蛋白ptm的差异,从而实现非常不同的寿命,展示了这一管道的实用性。©2020作者。基本方案1:同步秀丽隐杆线虫的大规模生长和收获。基本方案2:核制备、组蛋白提取和组蛋白纯化。基本方案3:组蛋白PTMs和组蛋白变体的自下而上质谱分析
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Current Protocols in Protein Science
Biochemistry, Genetics and Molecular Biology-Biochemistry
自引率
0.00%
发文量
0
期刊介绍:
With the mapping of the human genome, more and more researchers are exploring protein structures and functions in living organisms. Current Protocols in Protein Science provides protein scientists, biochemists, molecular biologists, geneticists, and others with the first comprehensive suite of protocols for this growing field.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


