Felipe A Louza, Guilherme P Telles, Simon Gog, Nicola Prezza, Giovanna Rosone
{"title":"gsufsort: constructing suffix arrays, LCP arrays and BWTs for string collections.","authors":"Felipe A Louza, Guilherme P Telles, Simon Gog, Nicola Prezza, Giovanna Rosone","doi":"10.1186/s13015-020-00177-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The construction of a suffix array for a collection of strings is a fundamental task in Bioinformatics and in many other applications that process strings. Related data structures, as the Longest Common Prefix array, the Burrows-Wheeler transform, and the document array, are often needed to accompany the suffix array to efficiently solve a wide variety of problems. While several algorithms have been proposed to construct the suffix array for a single string, less emphasis has been put on algorithms to construct suffix arrays for string collections.</p><p><strong>Result: </strong>In this paper we introduce gsufsort, an open source software for constructing the suffix array and related data indexing structures for a string collection with <i>N</i> symbols in <i>O</i>(<i>N</i>) time. Our tool is written in ANSI/C and is based on the algorithm gSACA-K (Louza et al. in Theor Comput Sci 678:22-39, 2017), the fastest algorithm to construct suffix arrays for string collections. The tool supports large fasta, fastq and text files with multiple strings as input. Experiments have shown very good performance on different types of strings.</p><p><strong>Conclusions: </strong>gsufsort is a fast, portable, and lightweight tool for constructing the suffix array and additional data structures for string collections.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"15 ","pages":"18"},"PeriodicalIF":1.5000,"publicationDate":"2020-09-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-020-00177-y","citationCount":"11","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-020-00177-y","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 11
Abstract
Background: The construction of a suffix array for a collection of strings is a fundamental task in Bioinformatics and in many other applications that process strings. Related data structures, as the Longest Common Prefix array, the Burrows-Wheeler transform, and the document array, are often needed to accompany the suffix array to efficiently solve a wide variety of problems. While several algorithms have been proposed to construct the suffix array for a single string, less emphasis has been put on algorithms to construct suffix arrays for string collections.
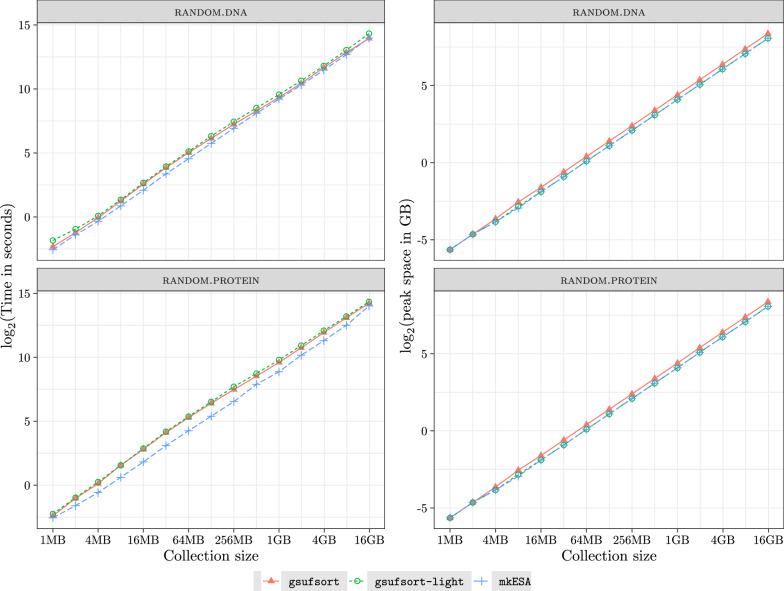
Result: In this paper we introduce gsufsort, an open source software for constructing the suffix array and related data indexing structures for a string collection with N symbols in O(N) time. Our tool is written in ANSI/C and is based on the algorithm gSACA-K (Louza et al. in Theor Comput Sci 678:22-39, 2017), the fastest algorithm to construct suffix arrays for string collections. The tool supports large fasta, fastq and text files with multiple strings as input. Experiments have shown very good performance on different types of strings.
Conclusions: gsufsort is a fast, portable, and lightweight tool for constructing the suffix array and additional data structures for string collections.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


