A linear-time algorithm that avoids inverses and computes Jackknife (leave-one-out) products like convolutions or other operators in commutative semigroups.
John L Spouge, Joseph M Ziegelbauer, Mileidy Gonzalez
{"title":"A linear-time algorithm that avoids inverses and computes Jackknife (leave-one-out) products like convolutions or other operators in commutative semigroups.","authors":"John L Spouge, Joseph M Ziegelbauer, Mileidy Gonzalez","doi":"10.1186/s13015-020-00178-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Data about herpesvirus microRNA motifs on human circular RNAs suggested the following statistical question. Consider independent random counts, not necessarily identically distributed. Conditioned on the sum, decide whether one of the counts is unusually large. Exact computation of the p<i>-</i>value leads to a specific algorithmic problem. Given <math><mi>n</mi></math> elements <math> <mrow><msub><mi>g</mi> <mn>0</mn></msub> <mo>,</mo> <msub><mi>g</mi> <mn>1</mn></msub> <mo>,</mo> <mo>…</mo> <mo>,</mo> <msub><mi>g</mi> <mrow><mi>n</mi> <mo>-</mo> <mn>1</mn></mrow> </msub> </mrow> </math> in a set <math><mi>G</mi></math> with the closure and associative properties and a commutative product without inverses, compute the jackknife (leave-one-out) products <math> <mrow> <msub> <mover><mrow><mi>g</mi></mrow> <mrow><mo>¯</mo></mrow> </mover> <mi>j</mi></msub> <mo>=</mo> <msub><mi>g</mi> <mn>0</mn></msub> <msub><mi>g</mi> <mn>1</mn></msub> <mo>⋯</mo> <msub><mi>g</mi> <mrow><mi>j</mi> <mo>-</mo> <mn>1</mn></mrow> </msub> <msub><mi>g</mi> <mrow><mi>j</mi> <mo>+</mo> <mn>1</mn></mrow> </msub> <mo>⋯</mo> <msub><mi>g</mi> <mrow><mi>n</mi> <mo>-</mo> <mn>1</mn></mrow> </msub> </mrow> </math> ( <math><mrow><mn>0</mn> <mo>≤</mo> <mi>j</mi> <mo><</mo> <mi>n</mi></mrow> </math> ).</p><p><strong>Results: </strong>This article gives a linear-time Jackknife Product algorithm. Its upward phase constructs a standard segment tree for computing segment products like <math> <mrow><msub><mi>g</mi> <mfenced><mrow><mi>i</mi> <mo>,</mo> <mi>j</mi></mrow> </mfenced> </msub> <mo>=</mo> <msub><mi>g</mi> <mi>i</mi></msub> <msub><mi>g</mi> <mrow><mi>i</mi> <mo>+</mo> <mn>1</mn></mrow> </msub> <mo>⋯</mo> <msub><mi>g</mi> <mrow><mi>j</mi> <mo>-</mo> <mn>1</mn></mrow> </msub> </mrow> </math> ; its novel downward phase mirrors the upward phase while exploiting the symmetry of <math><msub><mi>g</mi> <mi>j</mi></msub> </math> and its complement <math> <msub> <mover><mrow><mi>g</mi></mrow> <mrow><mo>¯</mo></mrow> </mover> <mi>j</mi></msub> </math> . The algorithm requires storage for <math><mrow><mn>2</mn> <mi>n</mi></mrow> </math> elements of <math><mi>G</mi></math> and only about <math><mrow><mn>3</mn> <mi>n</mi></mrow> </math> products. In contrast, the standard segment tree algorithms require about <math><mi>n</mi></math> products for construction and <math> <mrow><msub><mo>log</mo> <mn>2</mn></msub> <mi>n</mi></mrow> </math> products for calculating each <math> <msub> <mover><mrow><mi>g</mi></mrow> <mrow><mo>¯</mo></mrow> </mover> <mi>j</mi></msub> </math> , i.e., about <math><mrow><mi>n</mi> <msub><mo>log</mo> <mn>2</mn></msub> <mi>n</mi></mrow> </math> products in total; and a naïve quadratic algorithm using <math><mrow><mi>n</mi> <mo>-</mo> <mn>2</mn></mrow> </math> element-by-element products to compute each <math> <msub> <mover><mrow><mi>g</mi></mrow> <mrow><mo>¯</mo></mrow> </mover> <mi>j</mi></msub> </math> requires <math><mrow><mi>n</mi> <mfenced><mrow><mi>n</mi> <mo>-</mo> <mn>2</mn></mrow> </mfenced> </mrow> </math> products.</p><p><strong>Conclusions: </strong>In the herpesvirus application, the Jackknife Product algorithm required 15 min; standard segment tree algorithms would have taken an estimated 3 h; and the quadratic algorithm, an estimated 1 month. The Jackknife Product algorithm has many possible uses in bioinformatics and statistics.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"15 ","pages":"17"},"PeriodicalIF":1.5000,"publicationDate":"2020-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-020-00178-x","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-020-00178-x","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
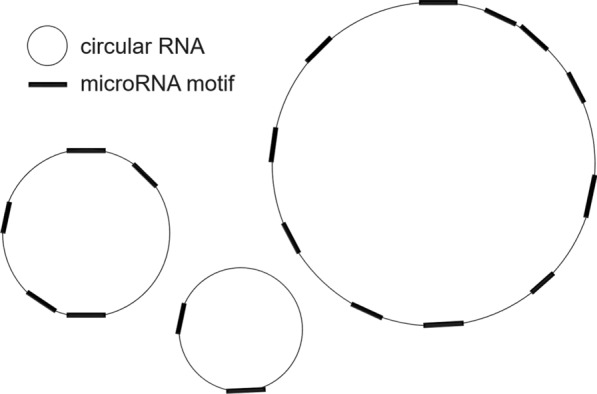
Background: Data about herpesvirus microRNA motifs on human circular RNAs suggested the following statistical question. Consider independent random counts, not necessarily identically distributed. Conditioned on the sum, decide whether one of the counts is unusually large. Exact computation of the p-value leads to a specific algorithmic problem. Given elements in a set with the closure and associative properties and a commutative product without inverses, compute the jackknife (leave-one-out) products ( ).
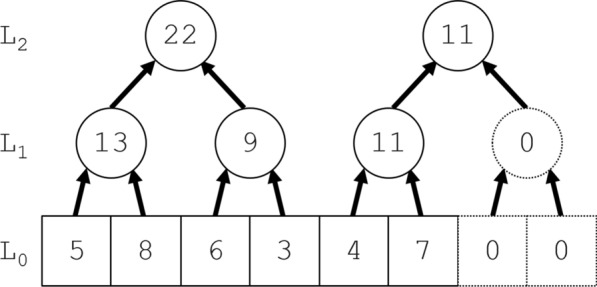
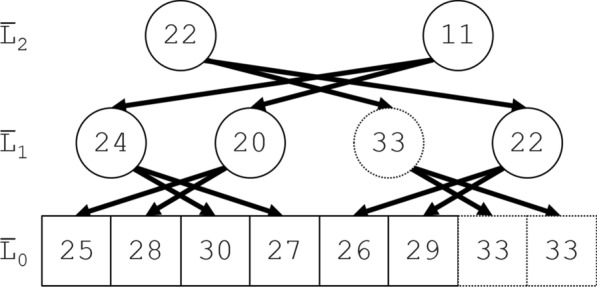
Results: This article gives a linear-time Jackknife Product algorithm. Its upward phase constructs a standard segment tree for computing segment products like ; its novel downward phase mirrors the upward phase while exploiting the symmetry of and its complement . The algorithm requires storage for elements of and only about products. In contrast, the standard segment tree algorithms require about products for construction and products for calculating each , i.e., about products in total; and a naïve quadratic algorithm using element-by-element products to compute each requires products.
Conclusions: In the herpesvirus application, the Jackknife Product algorithm required 15 min; standard segment tree algorithms would have taken an estimated 3 h; and the quadratic algorithm, an estimated 1 month. The Jackknife Product algorithm has many possible uses in bioinformatics and statistics.
背景:关于人环状rna上疱疹病毒microRNA基序的数据提出了以下统计学问题。考虑独立的随机计数,不一定是相同分布的。以总和为条件,决定其中一个计数是否异常大。p值的精确计算导致了一个特定的算法问题。给定集合g中的n个元素g 0, g 1,…,g n - 1,具有闭包性和结合性以及无逆的交换积,计算折刀(留一)积g¯j = g 0 g 1⋯g j - 1 g j + 1⋯g n - 1(0≤j n)。结果:给出了一种线性时间折刀积算法。它的向上阶段构建了一个标准的分段树,用于计算分段乘积,如g1, j = g i g i + 1⋯g j - 1;它新颖的向下相位反映了向上相位,同时利用了gj和它的补函数g¯j的对称性。该算法只需要存储2n个G元素和3n个乘积。相比之下,标准的段树算法需要大约n个乘积来构建,计算每个g¯j需要大约log 2n个乘积,即总共需要大约n log 2n个乘积;而naïve二次算法使用n - 2个逐元素乘积来计算每个g¯j需要n - n - 2个乘积。结论:在疱疹病毒应用中,Jackknife Product算法需要15 min;标准的片段树算法估计需要3小时;而二次算法,估计需要1个月。折刀产品算法在生物信息学和统计学中有许多可能的用途。
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


