Charlotte Hyldgaard, Janne Møller, Elisabeth Bendstrup
{"title":"Changes in management of idiopathic pulmonary fibrosis: impact on disease severity and mortality.","authors":"Charlotte Hyldgaard, Janne Møller, Elisabeth Bendstrup","doi":"10.1080/20018525.2020.1807682","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Idiopathic pulmonary fibrosis (IPF) is a serious interstitial lung disease (ILD) with a median survival of 3-5 years. The aim of the present study was to evaluate disease severity and survival in patients diagnosed with IPF in the era of antifibrotic therapies compared with an earlier IPF cohort.</p><p><strong>Methods: </strong>We identified all patients with fibrotic ILD in the hospital electronic case record system between 2011 and 2016, and reviewed each case in order to identify incident patients with IPF. We used the GAP-index to compare disease severity and mortality to previous findings in patients with IPF diagnosed at our center between 2003 and 2009.</p><p><strong>Results: </strong>260 patients were diagnosed with IPF between 2011 and 2016. Mean age was 72.6 years, 79% were male, mean forced vital capacity (FVC) was 80%, and mean diffusing capacity for carbon monoxide (DLco) was 44%. Age, FVC and DLco were significant predictors of mortality, but the presence of a typical usual interstitial pneumonia pattern on HRCT was not. Eighty percent of patients in GAP stage I received antifibrotic therapy, 73% in GAP stage II, and 29% in GAP stage III.The median survival was four years in the 2011-2016 cohort compared with three years in the 2003-2009 cohort. The distribution of patients between GAP stages was unchanged in 2011-2016 compared with 2003-2009, (stage I 34% vs. 32%, stage II 49% vs. 48% and stage III 20% vs. 16%). One-year mortality was 13% in 2011-2016 and 26% in 2003-2009. In severe disease (GAP stage III), one-year mortality was 26% and 54%, respectively, (p=0.019).</p><p><strong>Conclusion: </strong>Short-term mortality was significantly lower in the 2011-2016 cohort compared with 2003-2009. This improvement may be linked to changes in treatment strategies towards limited use of corticosteroids. Although early diagnosis of IPF still needs increased focus, the improvement is encouraging.</p>","PeriodicalId":11872,"journal":{"name":"European Clinical Respiratory Journal","volume":"7 1","pages":"1807682"},"PeriodicalIF":1.8000,"publicationDate":"2020-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/20018525.2020.1807682","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Clinical Respiratory Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/20018525.2020.1807682","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
引用次数: 5
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a serious interstitial lung disease (ILD) with a median survival of 3-5 years. The aim of the present study was to evaluate disease severity and survival in patients diagnosed with IPF in the era of antifibrotic therapies compared with an earlier IPF cohort.
Methods: We identified all patients with fibrotic ILD in the hospital electronic case record system between 2011 and 2016, and reviewed each case in order to identify incident patients with IPF. We used the GAP-index to compare disease severity and mortality to previous findings in patients with IPF diagnosed at our center between 2003 and 2009.
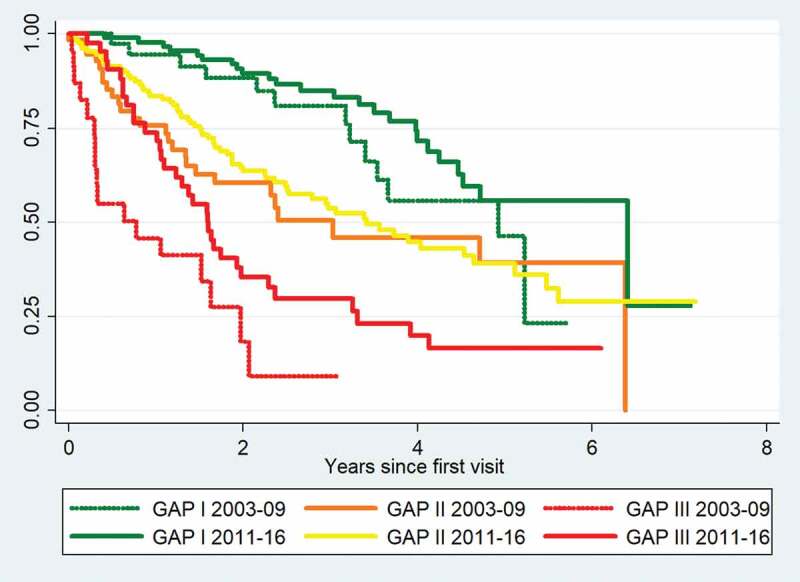
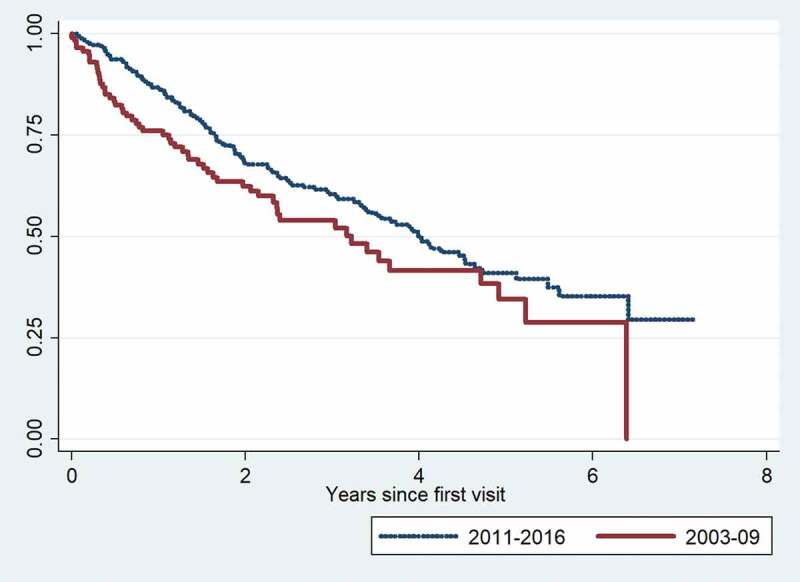
Results: 260 patients were diagnosed with IPF between 2011 and 2016. Mean age was 72.6 years, 79% were male, mean forced vital capacity (FVC) was 80%, and mean diffusing capacity for carbon monoxide (DLco) was 44%. Age, FVC and DLco were significant predictors of mortality, but the presence of a typical usual interstitial pneumonia pattern on HRCT was not. Eighty percent of patients in GAP stage I received antifibrotic therapy, 73% in GAP stage II, and 29% in GAP stage III.The median survival was four years in the 2011-2016 cohort compared with three years in the 2003-2009 cohort. The distribution of patients between GAP stages was unchanged in 2011-2016 compared with 2003-2009, (stage I 34% vs. 32%, stage II 49% vs. 48% and stage III 20% vs. 16%). One-year mortality was 13% in 2011-2016 and 26% in 2003-2009. In severe disease (GAP stage III), one-year mortality was 26% and 54%, respectively, (p=0.019).
Conclusion: Short-term mortality was significantly lower in the 2011-2016 cohort compared with 2003-2009. This improvement may be linked to changes in treatment strategies towards limited use of corticosteroids. Although early diagnosis of IPF still needs increased focus, the improvement is encouraging.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


