Case report: progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation.
Mariam Goubran, Ayodeji Aderibigbe, Emmanuel Jacquemin, Catherine Guettier, Safwat Girgis, Vincent Bain, Andrew L Mason
{"title":"Case report: progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation.","authors":"Mariam Goubran, Ayodeji Aderibigbe, Emmanuel Jacquemin, Catherine Guettier, Safwat Girgis, Vincent Bain, Andrew L Mason","doi":"10.1186/s12881-020-01173-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Progressive familial intrahepatic cholestasis (PFIC) type 3 is an autosomal recessive disorder arising from mutations in the ATP-binding cassette subfamily B member 4 (ABCB4) gene. This gene encodes multidrug resistance protein-3 (MDR3) that acts as a hepatocanalicular floppase that transports phosphatidylcholine from the inner to the outer canalicular membrane. In the absence of phosphatidylcholine, the detergent activity of bile salts is amplified and this leads to cholangiopathy, bile duct loss and biliary cirrhosis. Patients usually present in infancy or childhood and often progress to end-stage liver disease before adulthood.</p><p><strong>Case presentation: </strong>We report a 32-year-old female who required cadaveric liver transplantation at the age of 17 for cryptogenic cirrhosis. When the patient developed chronic ductopenia in the allograft 15 years later, we hypothesized that the patient's original disease was due to a deficiency of a biliary transport protein and the ductopenia could be explained by an autoimmune response to neoantigen that was not previously encountered by the immune system. We therefore performed genetic analyses and immunohistochemistry of the native liver, which led to a diagnosis of PFIC3. However, there was no evidence of humoral immune response to the MDR3 and therefore, we assumed that the ductopenia observed in the allograft was likely due to chronic rejection rather than autoimmune disease in the allograft.</p><p><strong>Conclusions: </strong>Teenage patients referred for liver transplantation with cryptogenic liver disease should undergo work up for PFIC3. An accurate diagnosis of PFIC 3 is key for optimal management, therapeutic intervention, and avoidance of complications before the onset of end-stage liver disease.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"238"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7708126/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01173-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
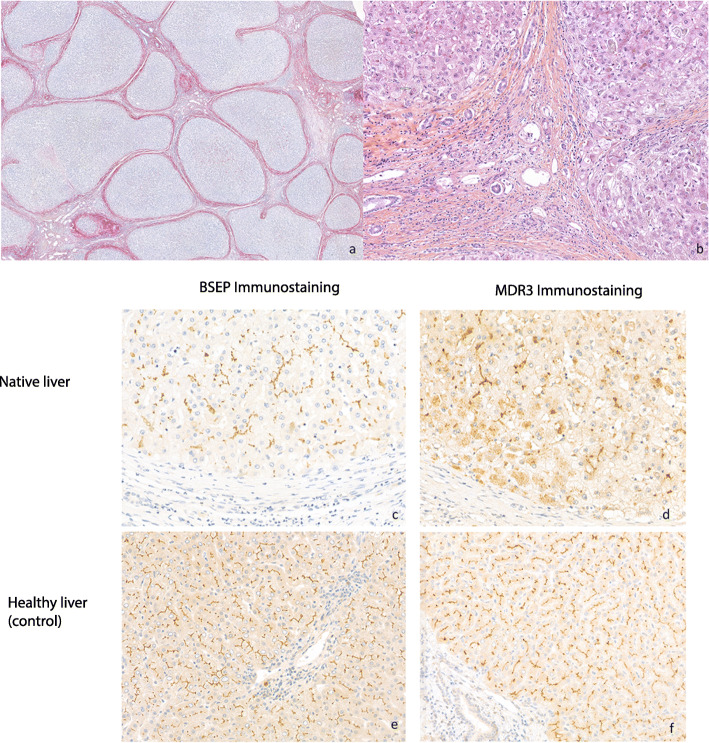
Background: Progressive familial intrahepatic cholestasis (PFIC) type 3 is an autosomal recessive disorder arising from mutations in the ATP-binding cassette subfamily B member 4 (ABCB4) gene. This gene encodes multidrug resistance protein-3 (MDR3) that acts as a hepatocanalicular floppase that transports phosphatidylcholine from the inner to the outer canalicular membrane. In the absence of phosphatidylcholine, the detergent activity of bile salts is amplified and this leads to cholangiopathy, bile duct loss and biliary cirrhosis. Patients usually present in infancy or childhood and often progress to end-stage liver disease before adulthood.
Case presentation: We report a 32-year-old female who required cadaveric liver transplantation at the age of 17 for cryptogenic cirrhosis. When the patient developed chronic ductopenia in the allograft 15 years later, we hypothesized that the patient's original disease was due to a deficiency of a biliary transport protein and the ductopenia could be explained by an autoimmune response to neoantigen that was not previously encountered by the immune system. We therefore performed genetic analyses and immunohistochemistry of the native liver, which led to a diagnosis of PFIC3. However, there was no evidence of humoral immune response to the MDR3 and therefore, we assumed that the ductopenia observed in the allograft was likely due to chronic rejection rather than autoimmune disease in the allograft.
Conclusions: Teenage patients referred for liver transplantation with cryptogenic liver disease should undergo work up for PFIC3. An accurate diagnosis of PFIC 3 is key for optimal management, therapeutic intervention, and avoidance of complications before the onset of end-stage liver disease.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


