{"title":"vcfView: An Extensible Data Visualization and Quality Assurance Platform for Integrated Somatic Variant Analysis.","authors":"Brian O'Sullivan, Cathal Seoighe","doi":"10.1177/1176935120972377","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Somatic mutations can have critical prognostic and therapeutic implications for cancer patients. Although targeted methods are often used to assay specific cancer driver mutations, high throughput sequencing is frequently applied to discover novel driver mutations and to determine the status of less-frequent driver mutations. The task of recovering somatic mutations from these data is nontrivial as somatic mutations must be distinguished from germline variants, sequencing errors, and other artefacts. Consequently, bioinformatics pipelines for recovery of somatic mutations from high throughput sequencing typically involve a large number of analytical choices in the form of quality filters.</p><p><strong>Results: </strong>We present vcfView, an interactive tool designed to support the evaluation of somatic mutation calls from cancer sequencing data. The tool takes as input a single variant call format (VCF) file and enables researchers to explore the impacts of analytical choices on the mutant allele frequency spectrum, on mutational signatures and on annotated somatic variants in genes of interest. It allows variants that have failed variant caller filters to be re-examined to improve sensitivity or guide the design of future experiments. It is extensible, allowing other algorithms to be incorporated easily.</p><p><strong>Availability: </strong>The shiny application can be downloaded from GitHub (https://github.com/BrianOSullivanGit/vcfView). All data processing is performed within <i>R</i> to ensure platform independence. The app has been tested on RStudio, version 1.1.456, with base <i>R</i> 3.6.2 and Shiny 1.4.0. A vignette based on a publicly available data set is also available on GitHub.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"19 ","pages":"1176935120972377"},"PeriodicalIF":2.4000,"publicationDate":"2020-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/1176935120972377","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/1176935120972377","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Motivation: Somatic mutations can have critical prognostic and therapeutic implications for cancer patients. Although targeted methods are often used to assay specific cancer driver mutations, high throughput sequencing is frequently applied to discover novel driver mutations and to determine the status of less-frequent driver mutations. The task of recovering somatic mutations from these data is nontrivial as somatic mutations must be distinguished from germline variants, sequencing errors, and other artefacts. Consequently, bioinformatics pipelines for recovery of somatic mutations from high throughput sequencing typically involve a large number of analytical choices in the form of quality filters.
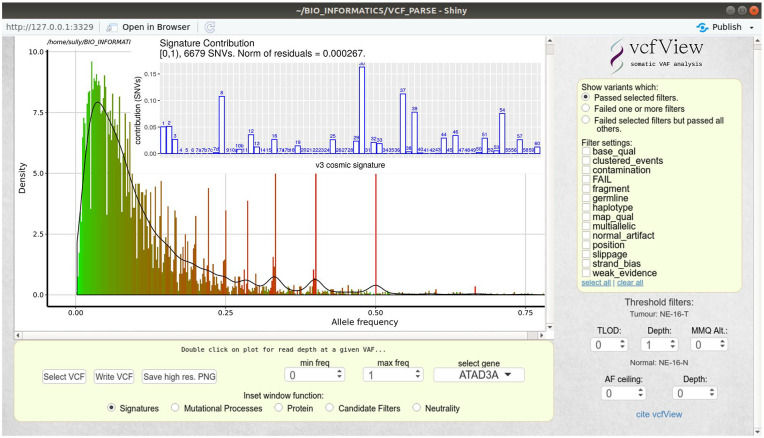
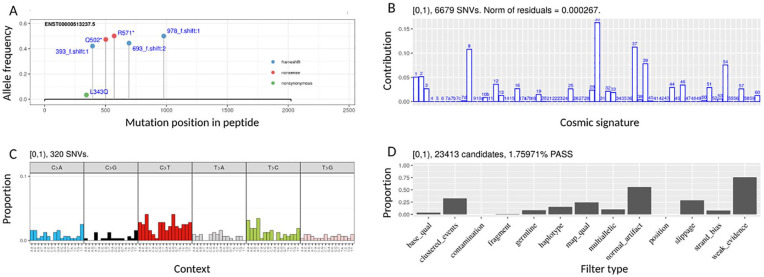
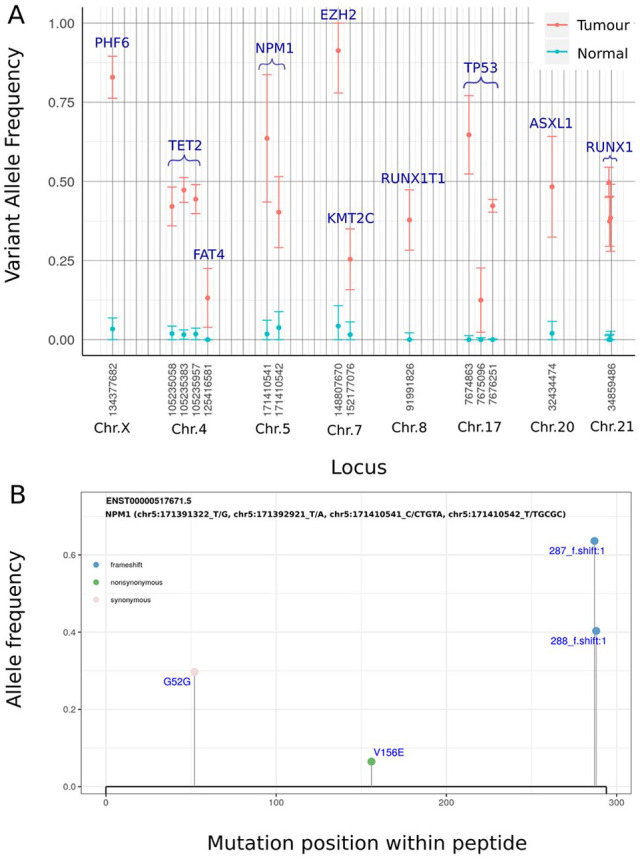
Results: We present vcfView, an interactive tool designed to support the evaluation of somatic mutation calls from cancer sequencing data. The tool takes as input a single variant call format (VCF) file and enables researchers to explore the impacts of analytical choices on the mutant allele frequency spectrum, on mutational signatures and on annotated somatic variants in genes of interest. It allows variants that have failed variant caller filters to be re-examined to improve sensitivity or guide the design of future experiments. It is extensible, allowing other algorithms to be incorporated easily.
Availability: The shiny application can be downloaded from GitHub (https://github.com/BrianOSullivanGit/vcfView). All data processing is performed within R to ensure platform independence. The app has been tested on RStudio, version 1.1.456, with base R 3.6.2 and Shiny 1.4.0. A vignette based on a publicly available data set is also available on GitHub.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


