Xiling Zhang, Zhimin Mao, Yanru Huang, Zhen Zhang, Jian Yao
{"title":"Gap junctions amplify TRPV4 activation-initiated cell injury via modification of intracellular Ca<sup>2+</sup> and Ca<sup>2+</sup>-dependent regulation of TXNIP.","authors":"Xiling Zhang, Zhimin Mao, Yanru Huang, Zhen Zhang, Jian Yao","doi":"10.1080/19336950.2020.1803552","DOIUrl":null,"url":null,"abstract":"<p><p>The elevated intracellular Ca<sup>2+</sup> and oxidative stress are well-reported mechanisms behind renal tubular epithelial injury initiated by various insults. Given that TRPV4 and connexin43 (Cx43) channels are activated by a wide range of stimuli and regulate both intracellular Ca<sup>2+</sup> and redox status, we speculated an involvement of these channels in renal tubular cell injury. Here, we tested this possibility and explored the potential underlying mechanisms. Our results demonstrated that exposure of renal tubular epithelial cells to aminoglycoside G418 led to cell death, which was attenuated by both TRPV4 and gap junction (Gj) inhibitor. Activation of TRPV4 caused cell damage, which was associated with an early increase in Cx43 expression and function. Inhibition of Cx43 with chemical inhibitor or siRNA largely prevented TRPV4 activation-induced cell damage. Further analysis revealed that TRPV4 agonists elicited a rise in intracellular Ca<sup>2+</sup> and caused a Ca<sup>2+</sup>-dependent elevation in TXNIP (a negative regulator of the antioxidant thioredoxin). In the presence of Gj inhibitor, however, these effects of TRPV4 were largely prevented. The depletion of intracellular Ca<sup>2+</sup> with Ca<sup>2+</sup> chelator BAPTA-AM or downregulation of TXNIP with siRNA significantly alleviated TRPV4 activation-initiated cell injury. Collectively, our results point to a critical involvement of TRPV4/Cx43 channel interaction in renal tubular cell injury through mechanisms involving a synergetic induction of intracellular Ca<sup>2+</sup> and oxidative stress. Channel interactions could be an important mechanism underlying cell injury. Targeting channels could have therapeutic potential for the treatment of acute tubular cell injury.</p>","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336950.2020.1803552","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2020.1803552","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 7
Abstract
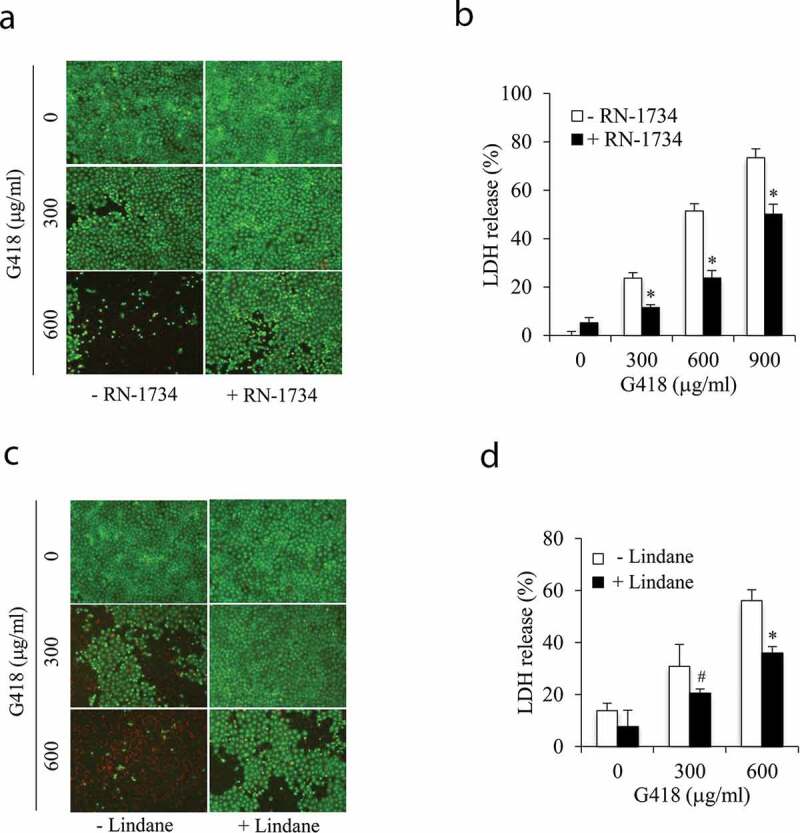
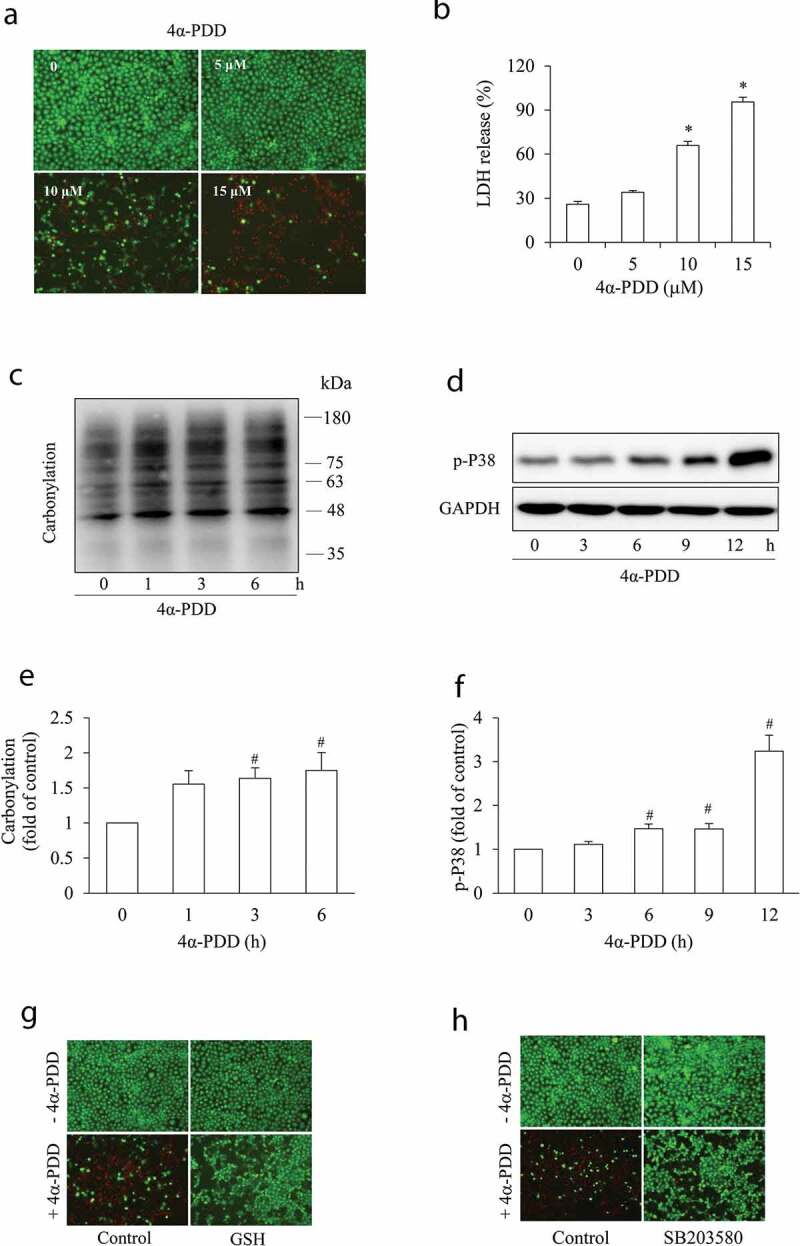
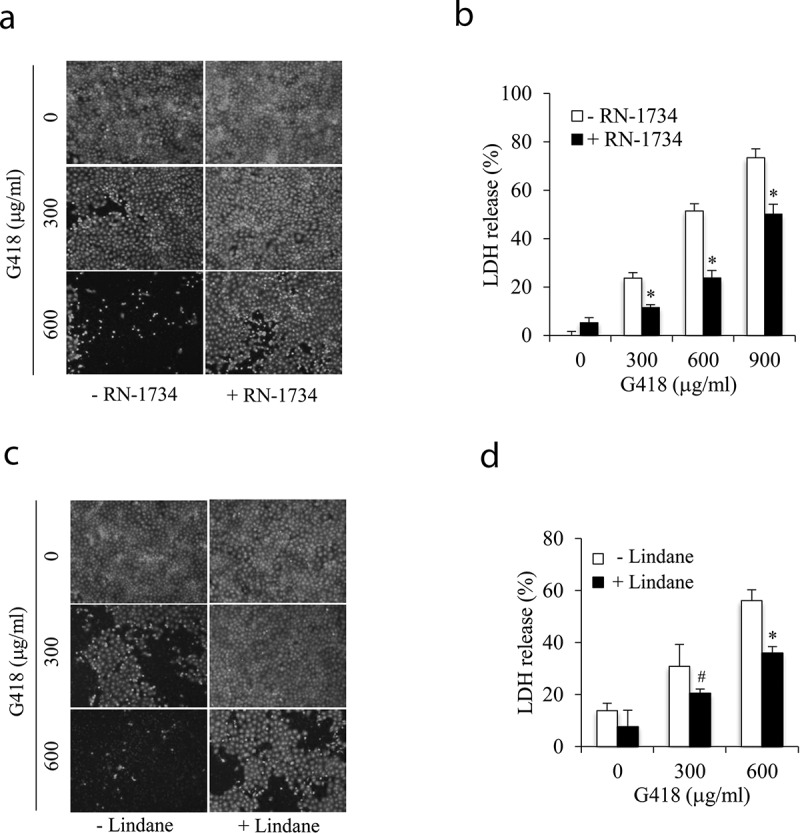
The elevated intracellular Ca2+ and oxidative stress are well-reported mechanisms behind renal tubular epithelial injury initiated by various insults. Given that TRPV4 and connexin43 (Cx43) channels are activated by a wide range of stimuli and regulate both intracellular Ca2+ and redox status, we speculated an involvement of these channels in renal tubular cell injury. Here, we tested this possibility and explored the potential underlying mechanisms. Our results demonstrated that exposure of renal tubular epithelial cells to aminoglycoside G418 led to cell death, which was attenuated by both TRPV4 and gap junction (Gj) inhibitor. Activation of TRPV4 caused cell damage, which was associated with an early increase in Cx43 expression and function. Inhibition of Cx43 with chemical inhibitor or siRNA largely prevented TRPV4 activation-induced cell damage. Further analysis revealed that TRPV4 agonists elicited a rise in intracellular Ca2+ and caused a Ca2+-dependent elevation in TXNIP (a negative regulator of the antioxidant thioredoxin). In the presence of Gj inhibitor, however, these effects of TRPV4 were largely prevented. The depletion of intracellular Ca2+ with Ca2+ chelator BAPTA-AM or downregulation of TXNIP with siRNA significantly alleviated TRPV4 activation-initiated cell injury. Collectively, our results point to a critical involvement of TRPV4/Cx43 channel interaction in renal tubular cell injury through mechanisms involving a synergetic induction of intracellular Ca2+ and oxidative stress. Channel interactions could be an important mechanism underlying cell injury. Targeting channels could have therapeutic potential for the treatment of acute tubular cell injury.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


