Elisia Clark, Joseph Johnson, Yi Na Dong, Elizabeth Mercado-Ayon, Nathan Warren, Mattieu Zhai, Emily McMillan, Amy Salovin, Hong Lin, David R Lynch
{"title":"Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease.","authors":"Elisia Clark, Joseph Johnson, Yi Na Dong, Elizabeth Mercado-Ayon, Nathan Warren, Mattieu Zhai, Emily McMillan, Amy Salovin, Hong Lin, David R Lynch","doi":"10.1042/NS20180060","DOIUrl":null,"url":null,"abstract":"<p><p>Friedreich ataxia (FRDA) is a progressive neurodegenerative disease with developmental features caused by a genetic deficiency of frataxin, a small, nuclear-encoded mitochondrial protein. Frataxin deficiency leads to impairment of iron-sulphur cluster synthesis, and consequently, ATP production abnormalities. Based on the involvement of such processes in FRDA, initial pathophysiological hypotheses focused on reactive oxygen species (ROS) production as a key component of the mechanism. With further study, a variety of other events appear to be involved, including abnormalities of mitochondrially related metabolism and dysfunction in mitochondrial biogenesis. Consequently, present therapies focus not only on free radical damage, but also on control of metabolic abnormalities and correction of mitochondrial biogenesis. Understanding the multitude of abnormalities in FRDA thus offers possibilities for treatment of this disorder.</p>","PeriodicalId":74287,"journal":{"name":"Neuronal signaling","volume":"2 4","pages":"NS20180060"},"PeriodicalIF":0.0000,"publicationDate":"2018-11-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1042/NS20180060","citationCount":"23","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuronal signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1042/NS20180060","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/12/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"Neuroscience","Score":null,"Total":0}
引用次数: 23
Abstract
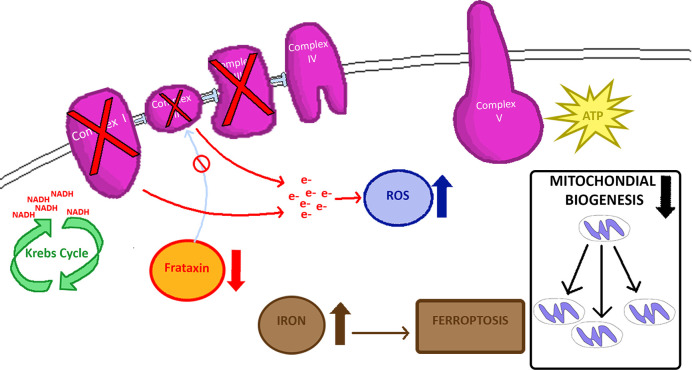
Friedreich ataxia (FRDA) is a progressive neurodegenerative disease with developmental features caused by a genetic deficiency of frataxin, a small, nuclear-encoded mitochondrial protein. Frataxin deficiency leads to impairment of iron-sulphur cluster synthesis, and consequently, ATP production abnormalities. Based on the involvement of such processes in FRDA, initial pathophysiological hypotheses focused on reactive oxygen species (ROS) production as a key component of the mechanism. With further study, a variety of other events appear to be involved, including abnormalities of mitochondrially related metabolism and dysfunction in mitochondrial biogenesis. Consequently, present therapies focus not only on free radical damage, but also on control of metabolic abnormalities and correction of mitochondrial biogenesis. Understanding the multitude of abnormalities in FRDA thus offers possibilities for treatment of this disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


