Helmuth-Günther Dörr, Nadja Schulze, Markus Bettendorf, Gerhard Binder, Walter Bonfig, Christian Denzer, Desiree Dunstheimer, Kirsten Salzgeber, Heinrich Schmidt, Karl Otfried Schwab, Egbert Voss, Martin Wabitsch, Joachim Wölfle
{"title":"Genotype-phenotype correlations in children and adolescents with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency.","authors":"Helmuth-Günther Dörr, Nadja Schulze, Markus Bettendorf, Gerhard Binder, Walter Bonfig, Christian Denzer, Desiree Dunstheimer, Kirsten Salzgeber, Heinrich Schmidt, Karl Otfried Schwab, Egbert Voss, Martin Wabitsch, Joachim Wölfle","doi":"10.1186/s40348-020-00100-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency is caused by mutations in the active 21-hydroxylase gene (CYP21A2). The clinical symptoms can vary greatly. To date, no systematic studies have been undertaken in Germany.</p><p><strong>Aims: </strong>Description of the phenotype, evaluation of the diagnostics and genotype-phenotype correlation PATIENTS AND METHODOLOGY: Retrospective analysis of the data of 134 patients (age range 0.1-18.6 years) in a multicentre study covering 10 paediatric endocrinology centres in Bavaria and Baden-Württemberg. The data was gathered on site from the medical records. Two hundred and thirty-three alleles with a mutation of the CYP21A2 gene were identified in 126 patients. A genotype-phenotype correlation of the mutation findings was undertaken (C1, severe/mild; C2, mild/mild). Individuals with a heterozygous mutation of the CYP21A2 were also included (C3). The data was collected with the approval of the ethics committee of the University Hospital of Erlangen during the period of 2014 and 2015. RESULTS (MW ± SD): One hundred and seventeen out of 134 patients (115 f, 29 m) were symptomatic. The chronological age (CA) at diagnosis was 7.1 ± 4.4 years. The most frequent symptom (73.5%) was premature pubarche. The height-SDS on diagnosis was 0.8 ± 1.3 and the BMI-SDS was 0.8 ± 1.2. Bone age (BA) was ascertained in 82.9% of the symptomatic patients. The difference between BA and CA was 1.9 ± 1.4 years. Basal 17OHP concentrations were 14.5 ± 19.1 ng/ml (18 patients < 2 ng/ml). In total, 58.1% mild and 34.7% severe mutations were found. The most common mutation was p.Val281Leu (39.1%); 65.8% of the patients could be allocated to group C1. No phenotypical differences were found between the 3 mutation groups. The 17OHP levels (basal and after ACTH) in the standard ACTH stimulation test were highest in group C1 and also significantly higher in group C2 as in C3, the ACTH-stimulated cortisol levels (ng/ml) were significantly lower in groups C1 (192.1 ± 62.5) and C2 (218 ± 50) than in C3 (297.3 ± 98.7).</p><p><strong>Conclusion: </strong>Most of the patients have symptoms of mild androgenisation. Male patients are underdiagnosed. Diagnostics are not standardised. Differences between the types of mutations are found in the hormone concentrations but not in phenotype. We speculate that further, as yet not clearly defined, factors are responsible for the development of the respective phenotypes.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"7 1","pages":"8"},"PeriodicalIF":3.4000,"publicationDate":"2020-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40348-020-00100-w","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-020-00100-w","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 6
Abstract
Background: Nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency is caused by mutations in the active 21-hydroxylase gene (CYP21A2). The clinical symptoms can vary greatly. To date, no systematic studies have been undertaken in Germany.
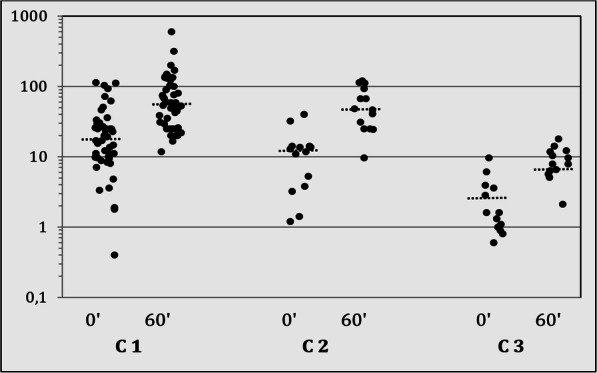
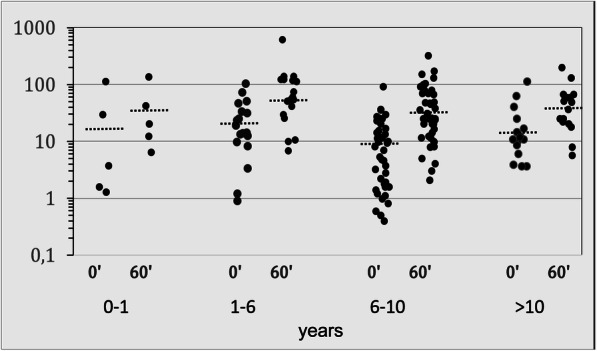
Aims: Description of the phenotype, evaluation of the diagnostics and genotype-phenotype correlation PATIENTS AND METHODOLOGY: Retrospective analysis of the data of 134 patients (age range 0.1-18.6 years) in a multicentre study covering 10 paediatric endocrinology centres in Bavaria and Baden-Württemberg. The data was gathered on site from the medical records. Two hundred and thirty-three alleles with a mutation of the CYP21A2 gene were identified in 126 patients. A genotype-phenotype correlation of the mutation findings was undertaken (C1, severe/mild; C2, mild/mild). Individuals with a heterozygous mutation of the CYP21A2 were also included (C3). The data was collected with the approval of the ethics committee of the University Hospital of Erlangen during the period of 2014 and 2015. RESULTS (MW ± SD): One hundred and seventeen out of 134 patients (115 f, 29 m) were symptomatic. The chronological age (CA) at diagnosis was 7.1 ± 4.4 years. The most frequent symptom (73.5%) was premature pubarche. The height-SDS on diagnosis was 0.8 ± 1.3 and the BMI-SDS was 0.8 ± 1.2. Bone age (BA) was ascertained in 82.9% of the symptomatic patients. The difference between BA and CA was 1.9 ± 1.4 years. Basal 17OHP concentrations were 14.5 ± 19.1 ng/ml (18 patients < 2 ng/ml). In total, 58.1% mild and 34.7% severe mutations were found. The most common mutation was p.Val281Leu (39.1%); 65.8% of the patients could be allocated to group C1. No phenotypical differences were found between the 3 mutation groups. The 17OHP levels (basal and after ACTH) in the standard ACTH stimulation test were highest in group C1 and also significantly higher in group C2 as in C3, the ACTH-stimulated cortisol levels (ng/ml) were significantly lower in groups C1 (192.1 ± 62.5) and C2 (218 ± 50) than in C3 (297.3 ± 98.7).
Conclusion: Most of the patients have symptoms of mild androgenisation. Male patients are underdiagnosed. Diagnostics are not standardised. Differences between the types of mutations are found in the hormone concentrations but not in phenotype. We speculate that further, as yet not clearly defined, factors are responsible for the development of the respective phenotypes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


