Predictive Metagenomic Profiling, Urine Metabolomics, and Human Marker Gene Expression as an Integrated Approach to Study Alopecia Areata.
Frontiers in Cellular and Infection Microbiology
Pub Date : 2020-04-29
eCollection Date: 2020-01-01
DOI:10.3389/fcimb.2020.00146
引用次数: 13
Abstract
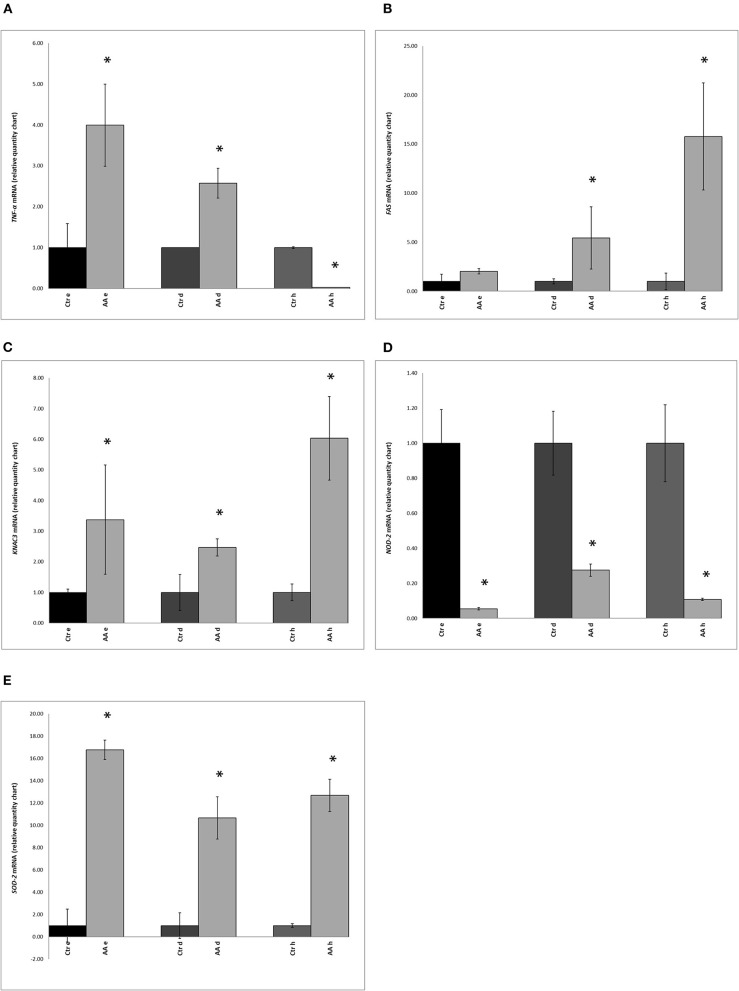
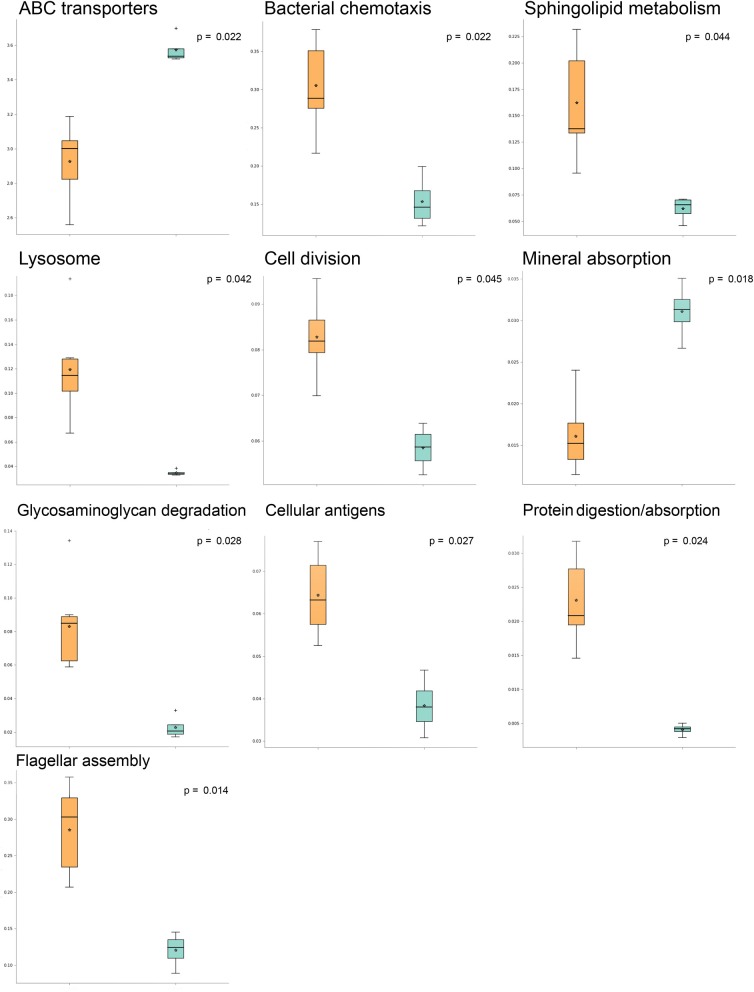
Involvement of the microbiome in many different scalp conditions has been investigated over the years. Studies on the role of the scalp microbiome in specific diseases, such as those involving hair growth alterations like non-cicatricial [androgenetic alopecia (AGA), alopecia areata (AA)] and cicatricial alopecia lichen planopilaris, are of major importance. In the present work, we highlighted the differences in microbial populations inhabiting the scalp of AA subjects and a healthy sample cohort by using an integrated approach relying on metagenomic targeted 16S sequencing analysis, urine metabolomics, and human marker gene expression. Significant differences in genera abundances (p < 0.05) were found in the hypodermis and especially the dermis layer. Based on 16S sequencing data, we explored the differences in predicted KEGG pathways and identified some significant differences in predicted pathways related to the AA pathologic condition such as flagellar, assembly, bacterial chemotaxis, mineral absorption, ABC transporters, cellular antigens, glycosaminoglycan degradation, lysosome, sphingolipid metabolism, cell division, protein digestion and absorption, and energy metabolism. All predicted pathways were significantly enhanced in AA samples compared to expression in healthy samples, with the exceptions of mineral absorption, and ABC transporters. We also determined the expression of TNF-α, FAS, KCNA3, NOD-2, and SOD-2 genes and explored the relationships between human gene expression levels and microbiome composition by Pearson's correlation analysis; here, significant correlations both positive (SOD vs. Staphylococcus, Candidatus Aquiluna) and negative (FAS and SOD2 vs. Anaerococcus, Neisseria, and Acinetobacter) were highlighted. Finally, we inspected volatile organic metabolite profiles in urinary samples and detected statistically significant differences (menthol, methanethiol, dihydrodehydro-beta-ionone, 2,5-dimethylfuran, 1,2,3,4, tetrahydro-1,5,7-trimethylnapthalene) when comparing AA and healthy subject groups. This multiple comparison approach highlighted potential traits associated with AA and their relationship with the microbiota inhabiting the scalp, opening up novel therapeutic interventions in such kind of hair growth disorders mainly by means of prebiotics, probiotics, and postbiotics.

预测性宏基因组分析、尿液代谢组学和人类标记基因表达作为研究斑秃的综合方法。
多年来,人们一直在研究微生物群在许多不同头皮状况中的作用。研究头皮微生物群在特定疾病中的作用,如涉及头发生长改变的疾病,如非瘢痕性[雄激素性脱发(AGA),斑秃(AA)]和瘢痕性脱发扁平苔藓,具有重要意义。在目前的工作中,我们利用基于宏基因组靶向16S测序分析、尿液代谢组学和人类标记基因表达的综合方法,强调了AA受试者和健康样本队列中生存的微生物种群的差异。属丰度差异有统计学意义(p < 0.05),真皮中属丰度差异最大。基于16S测序数据,我们探索了预测KEGG通路的差异,并发现与AA病理状况相关的鞭毛、组装、细菌趋化性、矿物质吸收、ABC转运蛋白、细胞抗原、糖胺聚糖降解、溶酶体、鞘脂代谢、细胞分裂、蛋白质消化吸收和能量代谢等预测通路存在显著差异。除了矿物质吸收和ABC转运蛋白外,AA样品中所有预测途径的表达都比健康样品显著增强。我们还检测了TNF-α、FAS、KCNA3、NOD-2和SOD-2基因的表达,并通过Pearson相关分析探讨了人类基因表达水平与微生物组组成之间的关系;在这里,显著的相关性(SOD与葡萄球菌、阿基卢纳候选菌)和负相关性(FAS和SOD2与厌氧球菌、奈瑟菌和不动杆菌)被强调。最后,我们检测了尿液样本中的挥发性有机代谢物谱,并在AA组与健康受试者组的比较中发现了具有统计学意义的差异(薄荷醇、甲硫醇、二氢脱氢- β -离子酮、2,5-二甲基呋喃、1,2,3,4、四氢-1,5,7-三甲基萘)。这种多重比较方法突出了与AA相关的潜在特征及其与头皮微生物群的关系,为主要通过益生元、益生菌和后益生菌治疗这类头发生长障碍开辟了新的治疗干预措施。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


