Tomasz Szmatoła, Artur Gurgul, Igor Jasielczuk, Weiwei Fu, Katarzyna Ropka-Molik
{"title":"A detailed characteristics of bias associated with long runs of homozygosity identification based on medium density SNP microarrays.","authors":"Tomasz Szmatoła, Artur Gurgul, Igor Jasielczuk, Weiwei Fu, Katarzyna Ropka-Molik","doi":"10.7150/jgen.39147","DOIUrl":null,"url":null,"abstract":"<p><p>In the present study, runs of homozygosity (ROH) detected with the use of a standard bovine 54k single nucleotide polymorphism (SNP) genotyping assay and two different ROH detection approaches, based on 50 (M1) or 15 (M2) consecutive SNPs, were compared with results of whole genome sequencing. Both microarray-based methods accurately recognised medium-sized ROH, however, it was found that M2 method seemed to better than M1 identify short ROH, but highly overestimated their number, leading to numerous false positive calls. Moreover, long ROH identified with microarray data tended to break into shorter segments in sequencing data because of the presence of regions with high heterozygosity within the ROH sequences. This may indicate, that these long ROH are formed by closely positioned shorter homozygous segments that may be of older origin or may be created by two similar but not identical haplotypes, showing minor internal recombination signs. Such finding also suggests that at least some of the results of previous studies in regard to long ROH may be biased leading to inaccurate estimations of genomes autozygosity via ROH classification into length categories.</p>","PeriodicalId":15834,"journal":{"name":"Journal of Genomics","volume":"8 ","pages":"43-48"},"PeriodicalIF":0.0000,"publicationDate":"2020-04-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.7150/jgen.39147","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.7150/jgen.39147","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 3
Abstract
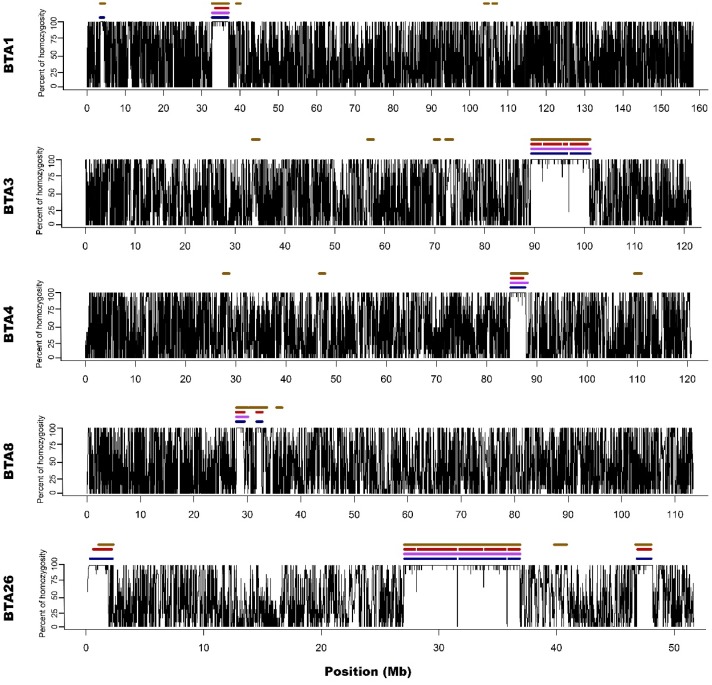
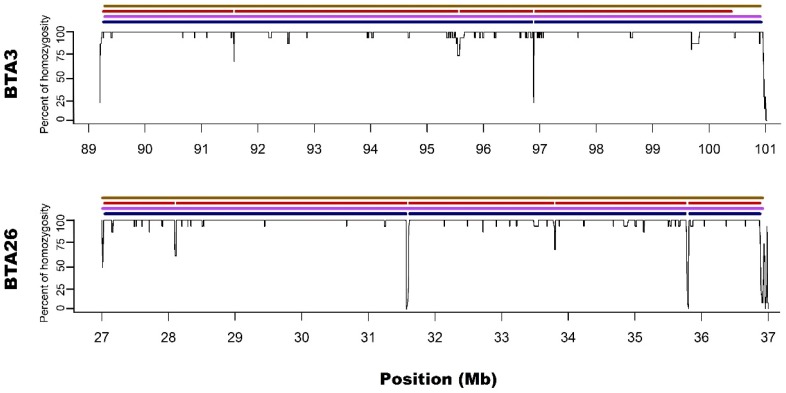
In the present study, runs of homozygosity (ROH) detected with the use of a standard bovine 54k single nucleotide polymorphism (SNP) genotyping assay and two different ROH detection approaches, based on 50 (M1) or 15 (M2) consecutive SNPs, were compared with results of whole genome sequencing. Both microarray-based methods accurately recognised medium-sized ROH, however, it was found that M2 method seemed to better than M1 identify short ROH, but highly overestimated their number, leading to numerous false positive calls. Moreover, long ROH identified with microarray data tended to break into shorter segments in sequencing data because of the presence of regions with high heterozygosity within the ROH sequences. This may indicate, that these long ROH are formed by closely positioned shorter homozygous segments that may be of older origin or may be created by two similar but not identical haplotypes, showing minor internal recombination signs. Such finding also suggests that at least some of the results of previous studies in regard to long ROH may be biased leading to inaccurate estimations of genomes autozygosity via ROH classification into length categories.
期刊介绍:
Journal of Genomics publishes papers of high quality in all areas of gene, genetics, genomics, proteomics, metabolomics, DNA/RNA, computational biology, bioinformatics, and other relevant areas of research and application. Articles published by the journal are rigorously peer-reviewed. Types of articles include: Research paper, Short research communication, Review or mini-reviews, Commentary, Database, Software.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


