Tzong-Shi Wang, Wen-Hsin Tsai, Li-Ping Tsai, Shi-Bing Wong
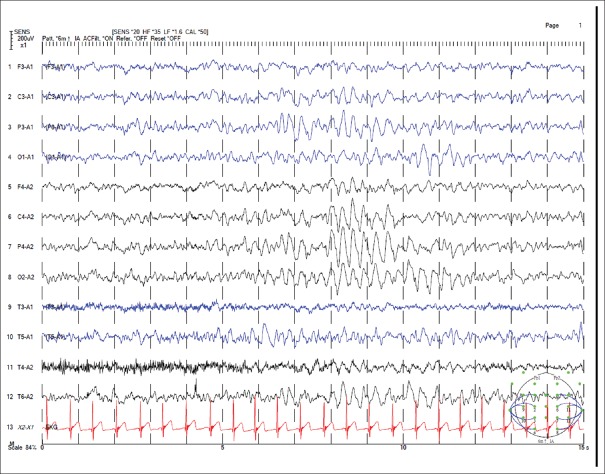
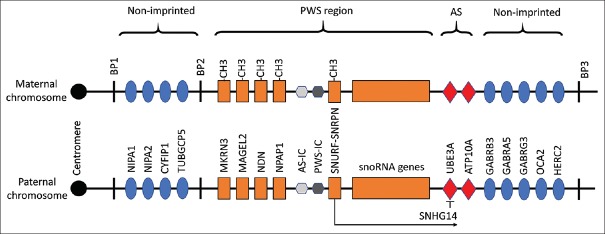
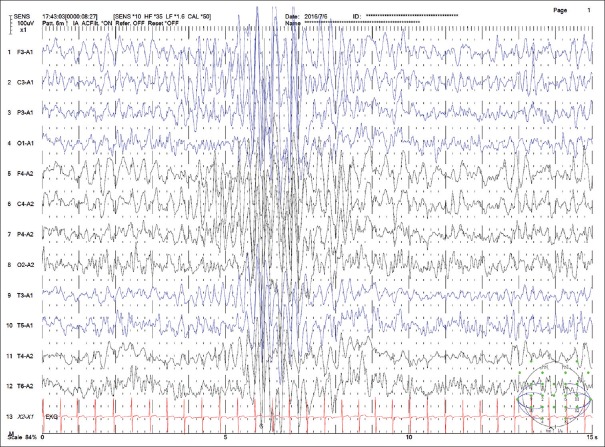
{"title":"Clinical characteristics and epilepsy in genomic imprinting disorders: Angelman syndrome and Prader-Willi syndrome.","authors":"Tzong-Shi Wang, Wen-Hsin Tsai, Li-Ping Tsai, Shi-Bing Wong","doi":"10.4103/tcmj.tcmj_103_19","DOIUrl":null,"url":null,"abstract":"<p><p>Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are considered sister imprinting disorders. Although both AS and PWS congenital neurodevelopmental disorders have chromosome 15q11.3-q13 dysfunction, their molecular mechanisms differ owing to genomic imprinting, which results in different parent-of-the-origin gene expressions. Recently, several randomized controlled trials have been proceeded to treat specific symptoms of AS and PWS. Due to the advance of clinical management, early diagnosis for patients with AS and PWS is important. PWS is induced by multiple paternal gene dysfunctions, including those in MKRN3, MAGEL2, NDN, SNURF-SNPRPN, NPAP1, and a cluster of small nucleolar RNA genes. PWS patients exhibit characteristic facial features, endocrinological, and behavioral phenotypes, including short and obese figures, hyperphagia, growth hormone deficiency, hypogonadism, autism, or obsessive- compulsive-like behaviors. In addition, hypotonia, poor feeding, failure to thrive, and typical facial features are major factors for early diagnosis of PWS. For PWS patients, epilepsy is not common and easy to treat. Conversely, AS is a single-gene disorder induced by ubiquitin-protein ligase E3A dysfunction, which only expresses from a maternal allele. AS patients develop epilepsy in their early lives and their seizures are difficult to control. The distinctive gait pattern, excessive laughter, and characteristic electroencephalography features, which contain anterior-dominated, high-voltage triphasic delta waves intermixed with epileptic spikes, result in early suspicion of AS. Often, polytherapy, including the combination of valproate, levetiracetam, lamotrigine, and benzodiazepines, is required for controlling seizures of AS patients. Notably, carbamazepine, oxcarbazepine, and vigabatrin should be avoided, since these may induce nonconvulsive status epilepticus. AS and PWS presented with distinct clinical manifestations according to specific molecular defects due to genomic imprinting. Early diagnosis and teamwork intervention, including geneticists, neurologists, rehabilitation physicians, and pulmonologists, are important. Epilepsy is common in patients with AS, and after proper treatment, seizures could be effectively controlled in late childhood or early adulthood for both AS and PWS patients.</p>","PeriodicalId":72593,"journal":{"name":"Ci ji yi xue za zhi = Tzu-chi medical journal","volume":"32 2","pages":"137-144"},"PeriodicalIF":0.0000,"publicationDate":"2019-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8a/2f/TCMJ-32-137.PMC7137370.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Ci ji yi xue za zhi = Tzu-chi medical journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4103/tcmj.tcmj_103_19","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/4/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are considered sister imprinting disorders. Although both AS and PWS congenital neurodevelopmental disorders have chromosome 15q11.3-q13 dysfunction, their molecular mechanisms differ owing to genomic imprinting, which results in different parent-of-the-origin gene expressions. Recently, several randomized controlled trials have been proceeded to treat specific symptoms of AS and PWS. Due to the advance of clinical management, early diagnosis for patients with AS and PWS is important. PWS is induced by multiple paternal gene dysfunctions, including those in MKRN3, MAGEL2, NDN, SNURF-SNPRPN, NPAP1, and a cluster of small nucleolar RNA genes. PWS patients exhibit characteristic facial features, endocrinological, and behavioral phenotypes, including short and obese figures, hyperphagia, growth hormone deficiency, hypogonadism, autism, or obsessive- compulsive-like behaviors. In addition, hypotonia, poor feeding, failure to thrive, and typical facial features are major factors for early diagnosis of PWS. For PWS patients, epilepsy is not common and easy to treat. Conversely, AS is a single-gene disorder induced by ubiquitin-protein ligase E3A dysfunction, which only expresses from a maternal allele. AS patients develop epilepsy in their early lives and their seizures are difficult to control. The distinctive gait pattern, excessive laughter, and characteristic electroencephalography features, which contain anterior-dominated, high-voltage triphasic delta waves intermixed with epileptic spikes, result in early suspicion of AS. Often, polytherapy, including the combination of valproate, levetiracetam, lamotrigine, and benzodiazepines, is required for controlling seizures of AS patients. Notably, carbamazepine, oxcarbazepine, and vigabatrin should be avoided, since these may induce nonconvulsive status epilepticus. AS and PWS presented with distinct clinical manifestations according to specific molecular defects due to genomic imprinting. Early diagnosis and teamwork intervention, including geneticists, neurologists, rehabilitation physicians, and pulmonologists, are important. Epilepsy is common in patients with AS, and after proper treatment, seizures could be effectively controlled in late childhood or early adulthood for both AS and PWS patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


