Md. Mehedi Hasan, Dianjing Guo and Hiroyuki Kurata
{"title":"Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information†","authors":"Md. Mehedi Hasan, Dianjing Guo and Hiroyuki Kurata","doi":"10.1039/C7MB00491E","DOIUrl":null,"url":null,"abstract":"<p >Cysteine S-sulfenylation is a major type of posttranslational modification that contributes to protein structure and function regulation in many cellular processes. Experimental identification of S-sulfenylation sites is challenging, due to the low abundance of proteins and the inefficient experimental methods. Computational identification of S-sulfenylation sites is an alternative strategy to annotate the S-sulfenylated proteome. In this study, a novel computational predictor SulCysSite was developed for accurate prediction of S-sulfenylation sites based on multiple sequence features, including amino acid index properties, binary amino acid codes, position specific scoring matrix, and compositions of profile-based amino acids. To learn the prediction model of SulCysSite, a random forest classifier was applied. The final SulCysSite achieved an AUC value of 0.819 in a 10-fold cross-validation test. It also exhibited higher performance than other existing computational predictors. In addition, the hidden and complex mechanisms were extracted from the predictive model of SulCysSite to investigate the understandable rules (<em>i.e.</em> feature combination) of S-sulfenylation sites. The SulCysSite is a useful computational resource for prediction of S-sulfenylation sites. The online interface and datasets are publicly available at http://kurata14.bio.kyutech.ac.jp/SulCysSite/.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 12","pages":" 2545-2550"},"PeriodicalIF":3.7430,"publicationDate":"2017-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C7MB00491E","citationCount":"48","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c7mb00491e","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 48
Abstract
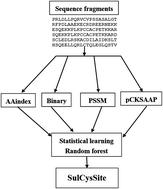
Cysteine S-sulfenylation is a major type of posttranslational modification that contributes to protein structure and function regulation in many cellular processes. Experimental identification of S-sulfenylation sites is challenging, due to the low abundance of proteins and the inefficient experimental methods. Computational identification of S-sulfenylation sites is an alternative strategy to annotate the S-sulfenylated proteome. In this study, a novel computational predictor SulCysSite was developed for accurate prediction of S-sulfenylation sites based on multiple sequence features, including amino acid index properties, binary amino acid codes, position specific scoring matrix, and compositions of profile-based amino acids. To learn the prediction model of SulCysSite, a random forest classifier was applied. The final SulCysSite achieved an AUC value of 0.819 in a 10-fold cross-validation test. It also exhibited higher performance than other existing computational predictors. In addition, the hidden and complex mechanisms were extracted from the predictive model of SulCysSite to investigate the understandable rules (i.e. feature combination) of S-sulfenylation sites. The SulCysSite is a useful computational resource for prediction of S-sulfenylation sites. The online interface and datasets are publicly available at http://kurata14.bio.kyutech.ac.jp/SulCysSite/.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


